原發惡性聽神經瘤1例及文獻回顧
2019-06-19 02:32:12蘇家豪李亮明林少華林其昌朱永華林偉標劉惠嬌
癌癥進展 2019年5期
蘇家豪,李亮明,林少華,林其昌,朱永華,林偉標,劉惠嬌
1中山大學附屬第三醫院神經外科,廣東中山510000
2中山市人民醫院神經外科,廣東中山528405
3中山大學附屬第一醫院重癥醫學科,廣東中山510000
惡性聽神經瘤是神經系統內罕見的惡性腫瘤[1],其發生于中樞神經系統第八對顱神經上,是一種極其罕見的惡性神經鞘瘤[2]。相關研究表明,惡性聽神經瘤通常繼發于腦部放療,以及聽神經鞘瘤γ刀治療后[3]。原發惡性聽神經瘤則更為罕見[4],國內僅報道過3例病例[5]。目前,原發惡性聽神經瘤的發病機制和臨床特征尚未明確[6]。本文對中山市人民醫院收治的1例原發惡性聽神經瘤患者的病歷資料進行回顧性分析,并結合相關文獻對原發惡性聽神經瘤患者的臨床特征和治療預后情況進行探討,現報道如下。
1 病歷資料
患者女,59歲,2017年11月23日因“右側面部麻木伴頭痛1月余”就診于中山市人民醫院,患者月前無明顯誘因出現右側面部麻木,伴頭痛。頭痛為陣發性,脹痛,自發緩解;面部麻木以右側Ⅱ、Ⅲ支明顯,無伴疼痛、抽搐,癥狀逐漸加重。患者既往史、個人史均未見異常。查體:右側面部感覺減退,Ⅱ、Ⅲ支明顯;聽力減退,右側明顯。電測聽示:右側輕度感音神經性耳聾。頭顱磁共振成像(magnetic resonance imaging,MRI)增強示:右側橋小腦角區占位,等T1稍長T2信號,明顯均勻強化大小為3.1 cm×2.9 cm×2.6 cm(圖1)。患者于2017年11月29日于中山市人民醫院行顯微手術下右側聽神經瘤切除術。術中見腫瘤實性、質軟,大小約3.1 cm×2.9 cm×2.6 cm,切開腫瘤包膜,先瘤內分塊切除腫瘤,見面神經位于腫瘤內下方,完整保留,常規磨除內聽道后壁,切除內聽道內腫瘤約0.5 cm×0.5 cm。術后中山市人民醫院病理診斷為惡性聽神經瘤,經中山大學附屬第一醫院病理科會診結果顯示:Ki-67陽性,約30%,細胞角蛋白(cytokeratin,CK)陰性,上皮細胞膜抗原(epithelial membrane antigen,EMA)陰性,結蛋白(Desmin)局灶陽性,平滑肌肌動蛋白(smooth muscle actin,SMA)局灶陽性,MyoD1陰性,S-100蛋白(S-100)部分陽性,膠質纖維酸性蛋白(glial fibrillary acidic protein,GFAP)陰性,P53陽性,波形蛋白(vimentin)陽性,CD34陽性,嗜鉻粒蛋白A(chromogranin A,CgA)陽性(右側聽神經瘤)(圖2)。確診為惡性聽神經瘤。

圖1 59歲女性原發惡性聽神經瘤患者的術前MRI圖像

圖2 59歲女性原發惡性聽神經瘤患者術后免疫組織化學結果(SP法,×200)
2017年12月6日,患者出院,出院時患者神清,無面癱,頭痛較前明顯緩解,傷口愈合良好。之后患者于中山市人民醫院放射科繼續治療,并定期復查。截至2018年3月6日,該患者隨訪過程中,復查MRI未見明顯復發征象(圖3)。查體:患者神清,右側輕度面癱,感覺減退,右耳耳聾,四肢活動正常,肌張力正常,腦膜刺激征陰性。
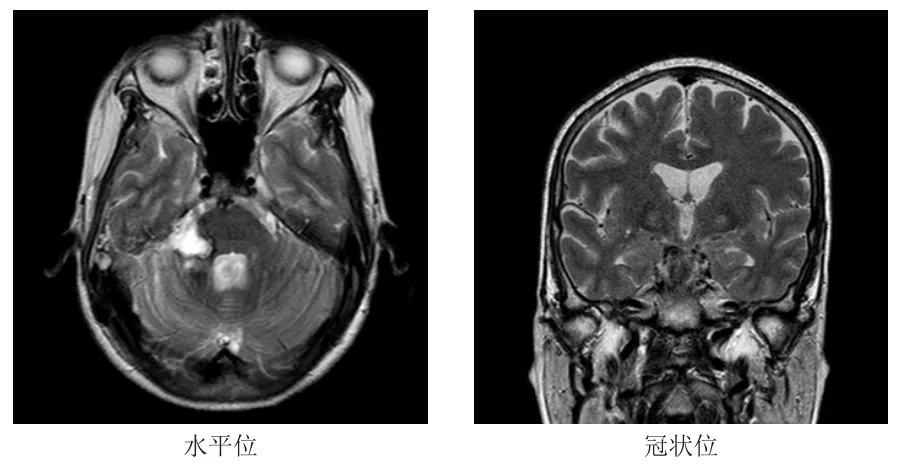
圖3 59歲女性原發性惡性聽神經瘤患者的術后復查MRI圖像
2 文獻檢索
以惡性聽神經瘤、神經鞘瘤惡性、惡性神經鞘瘤、第Ⅷ顱神經為中文檢索詞,以Malignant,acoustic neuroma,malignant schwannomas,VIII cranial nerve為英文檢索詞,單獨或組合的形式在PubMed、EMbase、Cochrane、中國生物醫學文獻數據庫(CBM)、維普(VIP)、中國期刊網全文數據庫(CNKI)、萬方數據庫進行檢索,收集1980年1月1日至2018年1月1日國內外公開發表的關于原發惡性聽神經瘤的文獻。經檢索共篩選出相關文獻15篇,共26例原發惡性聽神經瘤病例,其中國內文獻報道3例,國外文獻報道23例。
3 系統回顧
通過文獻檢索得到的26例原發惡性聽神經瘤病例和本病例,共納入27例原發惡性聽神經瘤患者,其中男12例,女15例;發病年齡為1.5~77.0歲,平均發病年齡為(42.87±19.88)歲;起病同聽神經鞘瘤,聽覺障礙最常見,為23例,伴隨毗鄰神經癥狀面癱14例,眩暈14例,腦積水3例,其他神經癥狀15例。在治療方面,所有患者均通過手術或術中活檢明確診斷,患者的中位生存時間為4個月。27例原發惡性聽神經瘤患者中,腫瘤全切11例,全切率為40.7%(11/27);術后復發16例,復發率為59.3%(16/27);術后放療8例,占29.6%(8/27);3例患者接受化療,其中僅1例患者生存[7-29]。(表1)
4 討論
惡性聽神經瘤是中樞神經系統極罕見的一種惡性腫瘤[30],目前對其尚缺乏大宗病例報道,國內外研究均以個案報道為主[31]。已有的研究報道顯示,惡性聽神經瘤的起病隱匿,其主要癥狀體征與聽神經鞘瘤無明顯區別[32],均表現為聽覺障礙、頭暈、腦積水、面癱[1];但是,惡性聽神經瘤的部分癥狀發生率比良性聽神經瘤高,如Ⅴ、Ⅵ、Ⅹ、Ⅺ等顱神經癥狀發生率較良性聽神經瘤高[31];而且,惡性聽神經瘤患者的腦積水、頭痛癥狀發生較早且發生率高于良性聽神經瘤患者[33]。臨床中,惡性聽神經瘤患者常被按照良性聽神經瘤進行治療,通常在腫瘤全切或部分切除后,經術后病理檢查得以確診,部分患者發現時腫瘤體積已經較大,手術全切較為困難,且術后并發癥多,為本病的治療帶來了一定困難[33]。本研究結果顯示,惡性聽神經瘤患者的預后較差,中位生存時間僅為4個月,1年生存率較低,且復發率較高,全切術后患者的1年內復發率達59.3%,值得神經外科醫師注意。

表1 1980年1月1日至2018年1月1日相關文獻中26例原發性惡性聽神經瘤患者及本例患者的病歷資料
目前,對惡性聽神經瘤及惡性神經鞘瘤的研究主要針對其組織起源及分子醫學特性展開[34]。相關文獻報道,原發性惡性聽神經瘤患者常伴發神經纖維瘤病(neurofibromatosis,NF)[3,35],但本例患者及國內報道病例中均未提及,考慮目前報道的病例較少,因此該觀點仍不能確定[2,36]。手術治療是臨床治療惡性聽神經瘤的主要手段。本文對本病例和相關文獻中的病例進行分析,結果發現27例原發惡性聽神經瘤患者中,11例患者行腫瘤全切手術,手術全切后患者的生存情況優于非全切及僅活檢的患者。由于目前病例數目較少,且考慮到部分切除患者往往由于腫瘤生長速度大且粘連明顯,導致切除困難,術后殘余多,因而部分切除患者的預后較全切除患者差,故單純評價手術全切對患者預后的影響尚不明確。本研究結果顯示,術后8例患者接受放療,放療劑量為50~60 Gy,術后放療患者的生存情況優于未行放療的患者。相關研究表明,惡性聽神經瘤患者對放療的反應性較好[37]。但目前惡性聽神經瘤病例較少且失訪率較高,各病例采用的放療方案及計劃均有差異,因此需要更多的數據支持[37]。目前尚無證據說明化療可以改善患者的預后,而對其他部位惡性聽神經瘤的治療中化療的應用也存在爭議[38]。
本研究通過原發惡性聽神經瘤的1例病例報告及對該病的系統回顧分析,對惡性聽神經瘤的臨床特征、治療手段及預后情況進行了分析。但是由于原發性惡性聽神經瘤的發病率極低,病例數少,治療方案及具體手段差異極大,對研究該病的臨床特征及預后帶來極大困難。
綜上所述,原發惡性聽神經瘤是一種極其罕見的中樞神經系統惡性腫瘤,其起病隱匿,癥狀與良性聽神經瘤相似,但惡性程度高,復發率和病死率均較高。目前病例分析發現,全切腫瘤及術后放療可以改善原發惡性聽神經瘤患者的預后,神經外科醫師在臨床工作中有必要提高對原發惡性聽神經瘤的認識,積極探索其綜合治療手段,改善患者預后。
