費-托蠟裂解混合α-烯烴齊聚制備潤滑油基礎油
2019-06-26 12:40:22劉東陽曹祖賓石薇薇李洪梅曹傳洋
石油化工 2019年6期
關鍵詞:催化劑
劉東陽,曹祖賓,石薇薇,李洪梅,曹傳洋
(1.遼寧石油化工大學 化學化工與環境學部,遼寧 撫順 113001;2.中國石油 撫順石化公司石油二廠,遼寧 撫順 113004)
聚α-烯烴(PAO)合成潤滑油基礎油是以α-烯烴為原料,在催化劑作用下聚合得到的多聚體混合物[1-2]。α-烯烴聚合催化劑包括路易斯酸催化劑、齊格勒-納塔催化劑、茂金屬催化劑和離子液體催化劑等[3-6]。α-烯烴的制備方法包括伯醇脫水法、費-托合成法、蠟裂解法和乙烯齊聚法等[7-10],工業上主要應用后兩種方法。乙烯齊聚法生產的1-癸烯是制備高性能PAO的主要單體,但1-癸烯的價格一直居高不下[11]。基于我國“富煤、缺油、少氣”的能源結構特點,采用煤基費-托蠟熱裂解生產α-烯烴在國內具有廣闊前景[12];但煤基費-托蠟熱裂解產物雜質多,直接以此為原料合成PAO的收率低、性能差[13]。
目前,關于蠟裂解α-烯烴提純的方法多為精餾切割和溶劑萃取,合成PAO收率較低。丁洪生等[14]對蠟裂解的混合α-烯烴進行精餾切割,得到C8~C13餾分,以此為原料進行了聚合研究。其中,α-烯烴含量為83.34%(w)時,合成的PAO在100 ℃的運動黏度為4.87 mm2/s。魚鯤等[15]采用無水三氯化鋁及白土吸附精制蠟裂解烯烴,精制后,α-烯烴純度為88.91%(w)。吳學謙等[16]采用二甲基亞砜為溶劑,對煤制烯烴進行精制處理,精制后的烯烴進行溶液聚合,合成的PAO在100 ℃的運動黏度為8.606 mm2/s。
本工作以費-托蠟裂解產物120~170 ℃餾分為原料,利用烯烴雙鍵與銀離子形成可逆的π絡合物的特點[17-18],對α-烯烴進行分離提純,得到高純度的α-烯烴,并對提純過程中形成的絡合物的組成進行了分析。采用BF3催化劑使提純后的α-烯烴齊聚,制備低黏度、高黏度指數的PAO,考察了單一變量對PAO性能的影響,并對PAO的組成分布、聚合過程中各官能團吸光度變化情況及產品支化度進行分析表征。
1 實驗部分
1.1 主要原料與試劑
N-甲基吡咯烷酮、硝酸銀、正丁醇、氫氧化鈉:分析純,國藥集團化學試劑有限公司;BF3:純度99.6%(w),山東省濱州市凱孚化工有限公司;費-托蠟:內蒙古伊泰煤制油有限公司。將費-托蠟在高壓反應釜中裂解制得液相產物,精餾切割120~170 ℃餾分為原料,產物組成分布見圖1。
1.2 α-烯烴分離提純
以硝酸銀為絡合劑,N-甲基吡咯烷酮為溶劑,配制濃度為3 mol/L的吸收液,將吸收液與費-托蠟裂解液按質量比為2∶1置于避光錐形瓶中,反應溫度為25 ℃,磁力攪拌2 h,靜置分層,上層無色透明液體為萃余相,下層萃取相為烯烴和銀離子形成的絡合物,呈淡黃色。將萃取相加水稀釋,升至50 ℃,磁力攪拌30 min,靜置分層,將絡合物解絡分離出α-烯烴,按式(1)計算α-烯烴的轉移率。


圖1 裂解產物組成分布Fig.1 Thermal cracking product composition distribution.
1.3 PAO合成
合成PAO的工藝流程見圖2。實驗開始前,用干燥的氮氣吹掃高壓反應釜,實驗時,在氮氣保護下向反應釜中加入定量混合的α-烯烴和正丁醇。升至反應溫度,開啟攪拌,轉速為200 r/min,向體系內通入BF3氣體至液面下,調節穩壓閥為一定值,使體系內BF3氣體維持一定壓力后,繼續通入BF3氣體催化劑,打開排氣閥,控制流量為6.5~7.0 L/h,尾氣接收裝置使用20%(w)的NaOH溶液吸收氣體。反應結束后,關停BF3,排空釜內壓力,取出聚合產物,加入5%(w)的NaOH溶液堿洗、水洗至中性后,減壓蒸餾除去280 ℃以下餾分,280 ℃及以上餾分作為目的產物。對聚合產物的黏度、傾點、閃點等性質進行測試,按式(2)計算PAO收率。

1.4 測試與表征
PAO的運動黏度按GB/T 265—1988[19]測試;黏度指數按GB/T 1995—1998[20]測試;密度按GB/T 4472—2011[21]測試;傾點按GB/T 3535—2006[22]測試;閃點按GB/T 3536—2008[23]測試;溴值按GB/T 11135—2013[24]測試;PAO的組成采用美國Agilent公司的7890B型高溫氣相色譜分析儀測試;分子結構采用Mettler Toledo公司的React IRTM4000型在線紅外分析儀測試;1H NMR采用布魯克拜厄斯賓有限公司的AVANCE Ⅲ HD 400型核磁共振波譜儀測試。PAO的支化度按式(3)計算。


圖2 合成PAO的工藝流程Fig.2 Process for synthesizing poly-α-olefin(PAO).
2 結果與討論
2.1 α-烯烴提純精制
2.1.1 絡合物組成
假設α-烯烴與銀離子形成絡合物的絡合比為1∶n,即一個α-烯烴分子絡合n個銀離子,形成的絡合物為(AO·nAg+)n+。α-烯烴絡合過程為:α-烯烴首先微溶于吸收液中,然后與吸收液中的銀離子形成絡合物,反應過程見式(4)~(5)。

式(4)達到平衡時,α-烯烴萃取相與萃余相間的分配系數按式(6)計算。

由式(5)可得絡合萃取平衡時的平衡常數,見式(7)。

α-烯烴總的絡合萃取平衡分配比按式(8)計算。
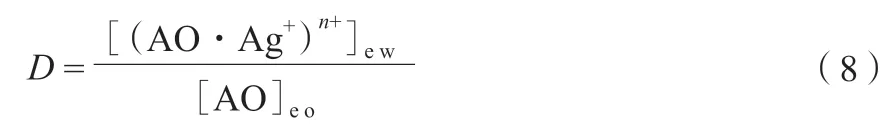
根據式(6)~(8),可推導出式(9)。

[Ag+]ew按式(10)計算。
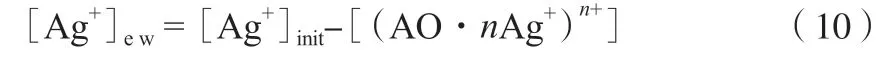
采用銀離子濃度不同的吸收液,對α-烯烴進行絡合萃取實驗,計算KD,K,D,利用絡合萃取實驗結果按式(9)得到lgD~lg[Ag+]ew擬合線,結果見圖3。由圖3可知,lgD與lg[Ag+]ew成線性關系,所得直線的斜率約為1,即n=1,說明生成的絡合物組成應為1∶1。這表明在實驗條件下1個α-烯烴分子絡合1個銀離子,即平均1個雙鍵絡合1個銀離子。

圖3 lgD~lg[Ag+]e w擬合關系曲線Fig.3 Fitting relationship curve of lgD-lg[Ag+]e w.
2.1.2 精制前后α-烯烴的組成分析
采用美國Agilent公司的7890A/5975C型氣質聯用儀對提純前后的α-烯烴組成進行分析,并按式(1)計算α-烯烴的轉移率,精制前后α-烯烴的純度及轉移率見表1。

表1 精制前后烯烴的純度及轉移率Table 1 Olefin purity and transfer rate before and after purification
由表1可知,精制后α-烯烴的純度由63.56%(w)增至95.25%(w),碳數分布為C7~C11,α-C10含量最高,為37.78%(w);α-烯烴轉移率為83.39%,α-C7轉移率最高,為91.14%(w);隨碳數增加,α-烯烴轉移率下降,說明隨碳鏈增加,銀離子絡合能力下降,因此,銀離子對低碳鏈α-烯烴絡合效果較好。
2.2 聚合工藝條件對PAO性能的影響
2.2.1 反應壓力
在反應溫度25 ℃、反應時間3 h、正丁醇質量分數為0.1%、BF3為催化劑的條件下,反應壓力對PAO收率和性能的影響見表2。采用BF3催化α-烯烴聚合,實質是催化劑與引發劑形成BF3-ROH(ROH:醇類化合物)型配位絡合物,產生活性中心,引發連鎖反應。由表2可知,當反應壓力較低時,反應釜內BF3催化劑濃度較低,絡合活性中心數目少,收率較低;隨著壓力增加,PAO收率增加,這是由于BF3分壓升高,溶解度變大,絡合活性中心數目增加,聚合能力提高,且壓力升高有利于聚合向正向移動;當壓力大于0.4 MPa時,PAO收率降低。因此,只有在適宜的壓力條件下,α-烯烴分子與正丁醇和BF3充分接觸,才能使PAO收率最高。壓力變化過程中,產品黏度、黏度指數出現先升高后降低趨勢,而傾點變化不明顯。綜合PAO收率和產品性能考慮,選取反應壓力為0.4 MPa。

表2 反應壓力對PAO收率和性能的影響Table 2 Effect of reaction pressure on PAO yield and performance
2.2.2 反應溫度
反應壓力0.4 MPa、反應時間3 h、正丁醇質量分數0.1%、BF3為催化劑的條件下,反應溫度對PAO收率和性能的影響見表3。由表3可知,隨著反應溫度升高,PAO收率、黏度和黏度指數降低,傾點升高,但在所考察溫度區間內,PAO收率均在90%以上。溫度不同,亨利系數不同,當壓強一定時,溫度升高,氣體運動速率加快,亨利系數增大,根據亨利定律, BF3氣體的飽和溶解度變小,使催化劑的有效濃度降低;且聚合為放熱反應,溫度升高,化學平衡向左移動,從而影響反應的轉化率;另一方面,低溫有利于高聚物的形成,高溫有利于低聚物的生成[25],溫度升高,聚合程度下降,表現為PAO運動黏度下降。反應溫度為10 ℃和25 ℃均可作為較佳反應溫度,為降低能耗,選取25 ℃為反應溫度。

表3 反應溫度對PAO收率和性能的影響Table 3 Effect of reaction temperature on PAO yield and performance
2.2.3 反應時間
在反應壓力0.4 MPa、反應溫度25 ℃、正丁醇質量分數為0.1%、BF3為催化劑的條件下,反應時間對PAO收率和性能的影響見表4。由表4可知,在反應時間小于3 h時,隨反應時間延長,PAO收率和運動黏度不斷增加,當反應時間大于3 h時,PAO收率和運動黏度增幅不大。這可能是由于經歷一定反應時間后,大部分α-烯烴形成了聚合物,PAO的性能趨于穩定,因此選取3 h作為反應時間。

表4 反應時間對PAO收率和性能的影響Table 4 Effect of reaction time on PAO yield and performance
2.2.4 正丁醇用量
在反應壓力0.4 MPa、反應溫度為25 ℃、反應時間為3 h、BF3為催化劑的條件下,正丁醇用量對PAO收率和性能的影響見表5。由表5可知,正丁醇用量由0.05%(w)增加到0.10%(w)過程中,PAO收率和黏度均呈現增加趨勢。這是由于引發劑用量增加,在引發劑作用下產生的活性中心數目增加,α-烯烴單體不斷被消耗,使PAO收率增加;但當引發劑用量過高時,活性中心濃度增大,聚合反應速率增加,發生異構化及分子內重排等副反應的幾率增大,影響產品性能,使黏度指數降低。綜合考慮PAO性能和收率,選取正丁醇質量分數為0.10%。
2.3 PAO性質及組成分析
2.3.1 PAO物化性質
以精制后混合α-烯烴為原料,在反應壓力0.4 MPa、反應溫度25 ℃、反應時間3 h、正丁醇質量分數為0.1%、BF3為催化劑的條件下進行聚合,聚合產物與商品PAO-6的物化性質列于表6[26]。由表6可知,合成的PAO在40,100 ℃的運動黏度分別為31.01,6.05 mm2/s,黏度指數為146,傾點為-62 ℃,具有很好的黏溫性能和低溫流動性能,各項性能均達到PAO-6的標準。

表5 正丁醇用量對PAO收率和性能的影響Table 5 Effect of n-butanol dosage on PAO yield and performance

表6 優化反應工藝條件下產品的性能Table 6 Performance of PAO product under the optimize reaction process conditions
2.3.2 組成分析
PAO的GC譜圖見圖4。由圖4可知,保留時 間 為6~10,10~15,15~23,23~26,26~29 min出現的峰分別對應PAO的二聚體、三聚體、四聚體、五聚體、六聚體,每組峰出現多個峰是由于產物中存在同分異構體,產物中聚合度大于6的多聚體含量較低。

圖4 PAO的GC譜圖Fig.4 GC spectrum of PAO.
用面積歸一法進行定量分析,得出各聚合物的組分含量,二聚體的含量為7.72%(w)、三聚體的含量為34.20%(w)、四聚體的含量為36.25%(w)、五聚體的含量為9.58%(w)、六聚體的含量為7.92%(w)、聚合度大于6的產物含量為4.33%(w),PAO產物中主要以三聚體和四聚體為主,含量為70.45%(w)。
2.4 在線紅外分析表征
2.4.1 齊聚反應不同時期紅外吸收光譜
將在線紅外分析系統探測器探頭放入到反應體系中,對合成過程中各官能團進行紅外光譜測定,不同時期的紅外吸收光譜見圖5。

圖5 不同時期的紅外吸收光譜Fig.5 Infrared absorption spectra of different periods.
由圖5可知,飽和烷烴的特征吸收峰主要有:3 000~2 800 cm-1為甲基和亞甲基伸縮振動吸收峰,1 460 cm-1處為亞甲基的變形振動吸收峰,1 375 cm-1處為甲基對稱變形振動吸收峰,720 cm-1處為亞甲基的平面搖擺振動吸收峰。烯烴的特征吸收峰主要出現在3 080,1 680,990,910 cm-1處。由圖5還可知,齊聚反應原料以及反應開始30 min的譜線中,雙鍵吸收峰略有下降,說明反應已經開始進行。與齊聚反應原料的譜線相比,反應結束時的譜線在3 080,990,910 cm-1處的吸收峰消失,說明聚合產物中不含烯烴;另外,在2 956,2 854,1 460,1 375,720 cm-1處出現強烈吸收峰,可知聚合產物為長鏈烷烴。
2.4.2 齊聚反應不同時期烯烴吸光度
3 080 cm-1處的吸收峰是=CH伸縮振動引起的,990 cm-1處的吸收峰歸屬于雙鍵在端基的烯烴(即α-烯烴)的伸縮振動。3 080,990 cm-1處的吸光度隨時間變化曲線見圖6。由圖6可知,990 cm-1處吸光度隨時間延長呈現下降趨勢,說明烯烴不斷減少。990 cm-1處吸光度隨時間延長平緩下降,而3 080 cm-1處的吸光度呈震蕩下降趨勢,可知混合α-烯烴在聚合過程中發生了歧化反應,生成了內烯烴,使反應體系中內烯烴濃度增加。根據Lambert-Beer定律,吸光度與濃度成正比,從而使3 080 cm-1處吸光度增加;隨之吸光度下降是由于生成的歧化產物也能參與烯烴齊聚反應而被消耗,生成鏈烷烴,從而出現=CH震蕩下降趨勢。齊聚反應至3 h時,吸光度隨時間變化曲線趨于平穩,說明聚合反應至3 h基本完成。

圖6 3 080,990 cm-1處的吸光度隨時間變化的曲線Fig.6 Curves of absorbance over time at 3 080 cm-1 and 990 cm-1.A3080:absorbance at 3 080 cm-1;A990:absorbance at 990 cm-1.
2.5 1H NMR分析
齊聚產物的1H NMR譜圖見圖7。由圖7可知,化學位移δ=0.5~1.0的吸收峰為甲基氫吸收峰,δ=1.0~2.0的吸收峰為亞甲基和次甲基氫吸收峰,可以判斷,合成潤滑油中甲基較少,亞甲基及次甲基較多,說明PAO主要由長鏈飽和烷烴組成。
通過計算1H NMR譜圖中各吸收峰的面積,得出合成PAO的甲基質子峰面積為1.00,亞甲基和次甲基質子峰面積為4.18。根據式(3)計算PAO支化度為0.159 5。合成PAO的支化度較低,表明具有良好的黏溫性能。

圖7 齊聚產物的1H NMR譜圖Fig.7 1H NMR spectrum of oligomerization products.
3 結論
1)采用絡合萃取法,對費-托蠟裂解產物的120~170 ℃餾分中的α-烯烴進行絡合萃取,形成的絡合物中α-烯烴與銀離子的絡合比為1∶1,精制后所得α-烯烴純度由63.56%(w)提高到95.25%(w),碳數分布為C7~C11。
2)以精制后的費-托蠟裂解液為原料,采用BF3為催化劑,在反應壓力0.4 MPa,反應溫度25℃,反應時間3 h,正丁醇質量分數為0.1%的條件下,合成的PAO在 40,100 ℃的運動黏度分別為31.01,6.05 mm2/s,黏度指數為146,傾點為-62℃,產品性能達到PAO-6質量標準。
3)費-托蠟裂解混合α-烯烴合成的PAO的聚合度較為集中,三聚體和四聚體含量為70.45%(w),主要成分為長鏈烷烴。
符號說明
ACH3甲基質子峰面積
ACH+CH2次甲基及亞甲基質子峰面積
AOα-烯烴
BI 支化度
D平衡分配比
K平衡常數
KD分配系數
m1萃取原料中α-烯烴的質量,g
m2萃取后α-烯烴的質量,g
m3聚合原料α-烯烴的總質量,g
m4≥280 ℃餾分的質量,g
n銀離子個數
δ轉移率,%
ηPAO收率,%
下角標
e 平衡狀態
init 原料
o 萃余相
w 萃取相
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
