多金屬氧酸鹽催化反應的研究進展
2019-07-05 08:44:26王遠超莊明妍田夢柯王皓月于曉晶
山東化工 2019年12期
關鍵詞:催化劑
王遠超,莊明妍,田夢柯,王皓月,于曉晶
(哈爾濱理工大學 化學與環境工程學院,黑龍江省高校綠色化工技術重點實驗室,黑龍江 哈爾濱 150040)
多金屬氧酸鹽(Polyoxometalates,POMs)由結構多樣的陽離子和聚陰離子簇組成,其中以MOx(x=5,6)的多金屬氧多面體為基本結構單元。在不同的條件下,聚陰離子扮演路易斯酸和路易斯堿的角色。此外,由于聚陰離子具有很強的攜電子和釋放電子的能力,常被認為是電子儲存體,表明其具有氧化還原性質。尤其是這些聚陰離子可通過人為設計獲得特殊性能,并對其結構和組成元素進行人工調整。催化劑在現代化學工業中占著及其重要的地位,為了尋找高效、綠色的催化劑,人們對催化劑的認識和研究不斷深入。
POMs不僅是一種新型綠色友好催化劑,同時還能催化多種類型的反應,比如氧化還原反應、烯烴聚合、酸催化、堿催化等,本文對其中一些反應進行了簡要概述。
1 氧化反應
POMs因其顯著的氧化還原性能、抗氧化劑的持久性和環境相容性而成為優勢催化劑。POMs與綠色氧化劑的組合不僅可以氧化烷烴、芳香烴甚至惰性脂肪族烷烴中的C原子,還可以氧化有機物中的S和N原子。
1.1 烯烴的環氧化
烯烴的初始氧化產物為環氧化物,水解后生成二元醇。Venturello-Ishii成功地利用POMs和H2O2實現對烯烴環氧化選擇性反應[1-2]。Venturello等首先以鎢酸鹽和磷酸鹽離子為催化劑前驅體,H2O2為供氧體,在兩相體系中進行烯烴環氧化反應。為了提高脂肪烯烴對水溶性催化劑前驅體的易接近性,將長鏈烷基修飾的季銨鹽(C6-C18)作為相轉移催化劑引入體系。在此反應條件下,鎢酸鹽與磷酸鹽反應生成過氧化物中間體{PO4[WO-(O2)2]4}3-,分離得到的過氧絡合物在類似條件下對烯烴的環氧化反應具有活性,因此被認為是活性的氧轉移物。
1.2 烷烴的氧化
1999年,Cavaleiro課題組[3]以H2O2為氧化劑,利用單取代TBA4H(CuPW11O39)和TBA4Hx[M(H2O)PW11O39](TBA4Hx(MPW11) (M=CoII,MnII,NiII,FeIII;n=5,4;x=1,0)氧化環己烷,其中TBA4(FePW11)具有更高的活性和反應選擇性。過量的H2O2對環己基過氧化物具有較高的選擇性。在TBA4(FePW11)存在下,使用過量的H2O2反應 2h后,可以實現74%的環辛烷轉化率和80%的環辛醇過氧化選擇性。
1.3 芳烴及其衍生物的氧化
POMs可催化芳烴及其衍生物的氧化獲得相應酚類、醌類等衍生物。有文獻證明H5V2PMo10可以相對活化芳烴低氧化電位,如蒽和4-甲氧基甲苯,最初通過自由基陽離子中間體的形成從碳氫化合物中提取一個電子,接下來是氧的轉移,從HPAs陰離子轉移到底物,形成氧合物[4-5]。在H5V2PMo10和1atm O2的作用下,333K 反應18h后,蒽的選擇性氧化(>99%)為蒽醌。
1.4 不飽和化合物的氧化鹵化
2003年,Firouzabadi等人[6]以H3PW12銫鹽為固體催化劑,實現了酚類及其他芳香族化合物的區域選擇性溴化反應,該鹽與十六烷基三甲基溴化銨(CTAB)結合,Br2為溴源催化反應。已知苯酚與Br2溴化反應生成多溴化產物,單溴化產物很難得到。然而,非均相Cs2.5H0.5PW12可以催化生成4-溴苯酚,分離收率94%。在二元催化體系中,CTAB對對溴化產物具有良好的區域選擇性,而Cs2.5H0.5PW12是提高了反應速率。
1.5 含硫化合物的氧化
Thorimbert等人將酰胺羰基插入Dawson型聚陰離子[V3P2W15O62]9-(V3P2W15)的V3P2W15球中[7]。研究發現,羰基的加入提高了硫化物氧化反應的化學選擇性,而不會使POMs的V核失去活性。在催化劑中引入具有路易斯酸性的Pd中心,得到雙位催化劑。Pd中心的引入保留了V的氧化性質。
2 酸催化反應
2.1 C-C鍵形成的反應
在甲苯與1-辛烯(方程式1)的液相付-克烷基化反應中,H4SiW12/MCM-41和H3PW12/MCM-41表現出比本體形式高得多的活性[8]。1-辛烯的轉化率接近100%,在393K下,H4SiW12/MCM-41催化反應2h,2-異構體單烷基化產物選擇性高達99.9%。納米Cs2.5H0.5PW12/K-10顆粒催化對茴香醚與環己烯的烷基化反應,生成2-環己烷唑和4-環己烷唑(方程式2)[9]。
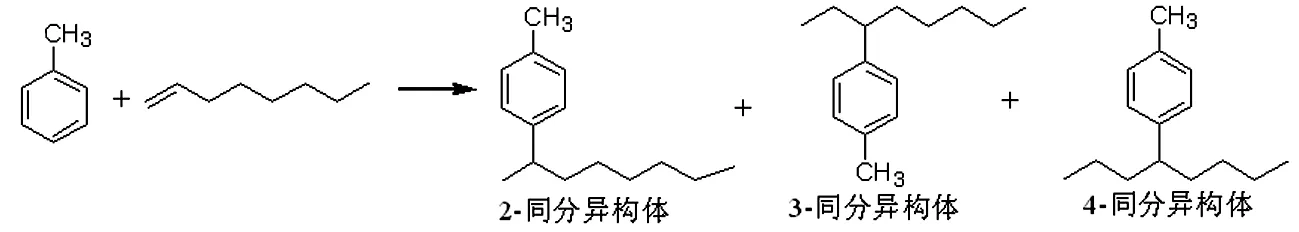
(方程式1)
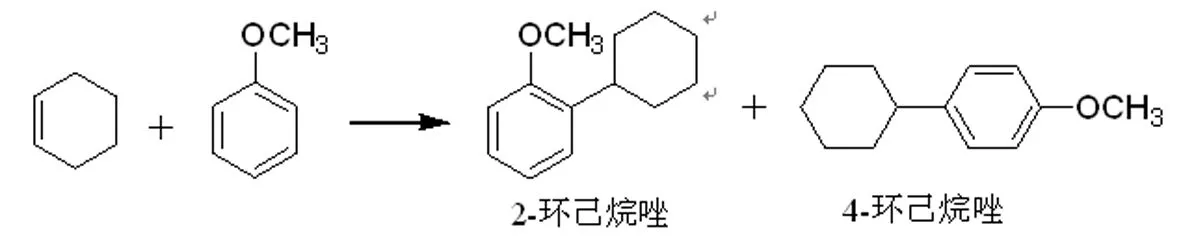
(方程式2)
2.2 C-X鍵形成的反應(X=N,S)
C-X鍵的形成(X=N,S)可用于生物活性和藥理活性復合物的合成。在H3PMo12或H3PW12存在下,用失活芳香胺對環氧化物進行氨解[10],生成相應的β-氨基醇,其產率較高(方程式3)。
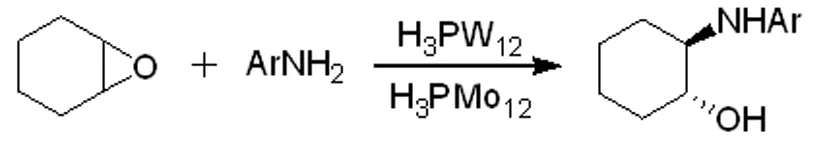
(方程式3)
C-S鍵的形成發生在芳烴和亞硫酰氯合成對稱二芳基亞砜(方程式4)和在H3PW12存在下芳基鹵化物和硫醇合成硫化物(方程式5)[11]的過程中。這兩種催化方法都具有反應時間短、產率高、環境條件好、操作簡單等優點。
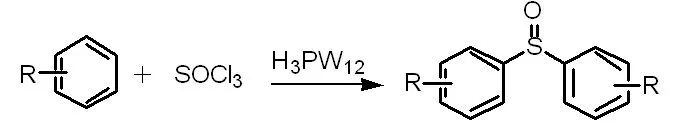
(方程式4)
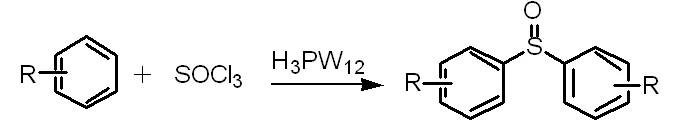
(方程式5)
2.3 磷酸酯的水解
2008年,Parac-Vogt課題組發現,帶高負電荷的七鉬酸鹽陰離子[Mo7O24]-6能夠有效催化DNA模型底物中常用的雙(4-硝基苯基)磷酸鹽(BNPP)中磷酸二酯鍵的裂解[12],與pH值為5.5時的非催化裂解速率相比,在[Mo7O24]-6上的反應速率加快了近4個數量級。與初始產物4-硝基苯基磷酸鹽(NPP)相比,BNPP中第一個磷酸酯鍵的裂解速度要慢得多。具體地說,相同條件下,NPP中磷酸二酯鍵的裂解速度比BNPP中第一個磷酸二酯鍵的裂解速度快近40倍。
3 有機疊氮環加成反應
2008年,Mizuno的小組報告了有機疊氮化物對炔的區域選擇性1,3-偶極環加成[13]。以往用于1,3-偶極環加成的銅催化劑幾乎都是單核的。TBA4(γ-H2Cu2SiW10)是第一個雙核銅反應催化劑。它的活性遠高于許多單核銅催化劑。
4 CO2環加成反應
2005年,Sakakura等人使用過渡金屬取代的 POMs[(n-C7H15)4N]x[α-M(H2O)SiW11O39](M=Co2+,Mn2+,Ni2+,Fe3+,Cu2+;x=5,6)用于CO2與1,2-環氧丙烷的反應[14](方程式6)。他們發現鈷和錳取代的催化劑在堿性和無鹵條件下的活性最高。
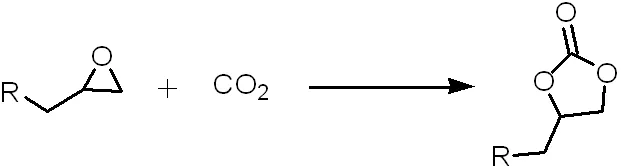
(方程式6)
5 耦聯反應
紐曼等人使用單鈀取代的POMs,K5[PdPW11O39]作為Suzuki、Heck和Stille型C-C耦聯和C-N耦聯反應的無機催化劑前體[15]。上述系統不需要額外的有機配體,因為Keggin骨架可以充當配體來穩定活性金屬中心。
6 結語
本文綜述了POMs催化的一些反應,包括氧化反應、酸催化、環加成反應和耦聯反應。這些例子也說明了在滿足不同反應要求的情況下,POMs的催化性能可以很好地調節,而且由于POMs具有多種催化功能和易于改性,很容易制備雙功能催化劑。特別是這些雙功能POMs使一些級聯反應在一鍋內順利進行,為有機合成提供了簡單的方案,這對于提高生產力和簡化整個反應程序是十分重要的。總之,POMs具有固有的穩定性、有效性和可設計性,這些優點使POMs催化劑成為工業催化劑的理想選擇。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
