一測多評法測定毛郁金中姜黃素和去甲氧基姜黃素含量*
2019-07-06 07:50:32劉偲翔蔣秀珍賴茂祥劉布鳴
中國藥業 2019年13期
關鍵詞:質量
劉偲翔 ,黃 艷 ,蔣秀珍 ,柴 玲 ,賴茂祥 ,林 霄 ,劉布鳴 △
(1.廣西中醫藥大學附屬瑞康醫院,廣西 南寧 530011; 2.廣西中醫藥研究院·廣西中藥質量標準研究重點實驗室,廣西 南寧 530022)
一測多評(QAMS)是一種用于多成分質量控制的分析方法,可解決中藥質量控制中對照品短缺問題,降低中藥質量控制分析成本。目前,該方法已成功應用于黃連、木通、人參等20多種常用中藥的質量控制評價。毛郁金為姜科植物毛郁金Curacuma aromaticaSalisb.的干燥根莖,其標準收載于《廣西壯族自治區壯藥質量標準·第二卷》[1],具有行氣解郁、涼血破瘀和利膽作用,用于治療胸悶脅痛、胃腹脹痛、黃疸、吐血、尿血、月經不調和癲癇等病癥[2]。藥理試驗證明,毛郁金具有鎮痛、止血[3]、抗炎[4]及免疫抑制[5]等作用。姜黃素類化合物是毛郁金藥材的主要活性成分,具有良好的抗氧化、抗炎、抗腫瘤、降血糖、抗HIV功效,可治療心血管疾病和阿爾茨海默病等[6]。本課題組前期研究中已分離得到毛郁金中姜黃素、去甲氧基姜黃素等17個化合物,其中大部分為倍半萜化合物[7],還含有揮發油等化合物[8-9],并采用高效液相色譜(HPLC)法建立了毛郁金中姜黃素的含量測定方法[10]。但僅以姜黃素為指標性成分對毛郁金藥材進行質量控制,且其含量較低,尚不能真正體現和保證毛郁金的內在本質及其相關制劑的質量。姜黃素和去甲氧基姜黃素為同一化學母核,在藥材中的相對含量相當,為了更好地保證毛郁金的有效性、安全性和專屬性,故以兩者為指標評價毛郁金質量。本研究中采用一測多評法測定了毛郁金中姜黃素和去甲氧基姜黃素的含量,以探討一測多評法在毛郁金多指標質量評價中的適用性和可行性。現報道如下。
1 儀器與試藥
儀器:KQ5200B型超聲波清洗器(昆山市超聲儀器有限公司);XS205型分析天平(梅特勒-托利多公司);UC3282型高效液相色譜儀,UC3292型紫外檢測儀,均購于南寧威瑪龍色譜科技有限公司;Waters1525-1998型高效液相色譜儀(沃特世公司);UV2550紫外可見分光光度儀(日本島津公司)。
試藥:姜黃素(純度 >98%,批號為 111530-201208),去甲氧基姜黃素對照品(純度>98%,批號為111531-201208),均購自中國食品藥品檢定研究院;乙腈(色譜級,賽默飛世爾科技有限公司),磷酸(分析純,廣州新建精細化工廠),其他試劑均為分析純;毛郁金Curacuma aromaticaSalisb.根莖采自廣西橫縣、靈山和南寧西郊,經廣西中醫藥研究院賴茂祥研究員鑒定為正品。
2 方法與結果
2.1 色譜條件與系統適用性試驗
色譜柱:Ecosil ODS-Extemnd柱(250 mm×4.6 mm,5 μm);流動相:乙腈 -4%磷酸溶液(42 ∶58,V∶V);流速:1.0 mL/min;檢測波長:425 nm;柱溫:25 ℃;進樣量:10μL。理論板數按姜黃素峰計算應不低于5000,在此色譜條件下,可完全分離。該色譜條件下色譜圖見圖1。
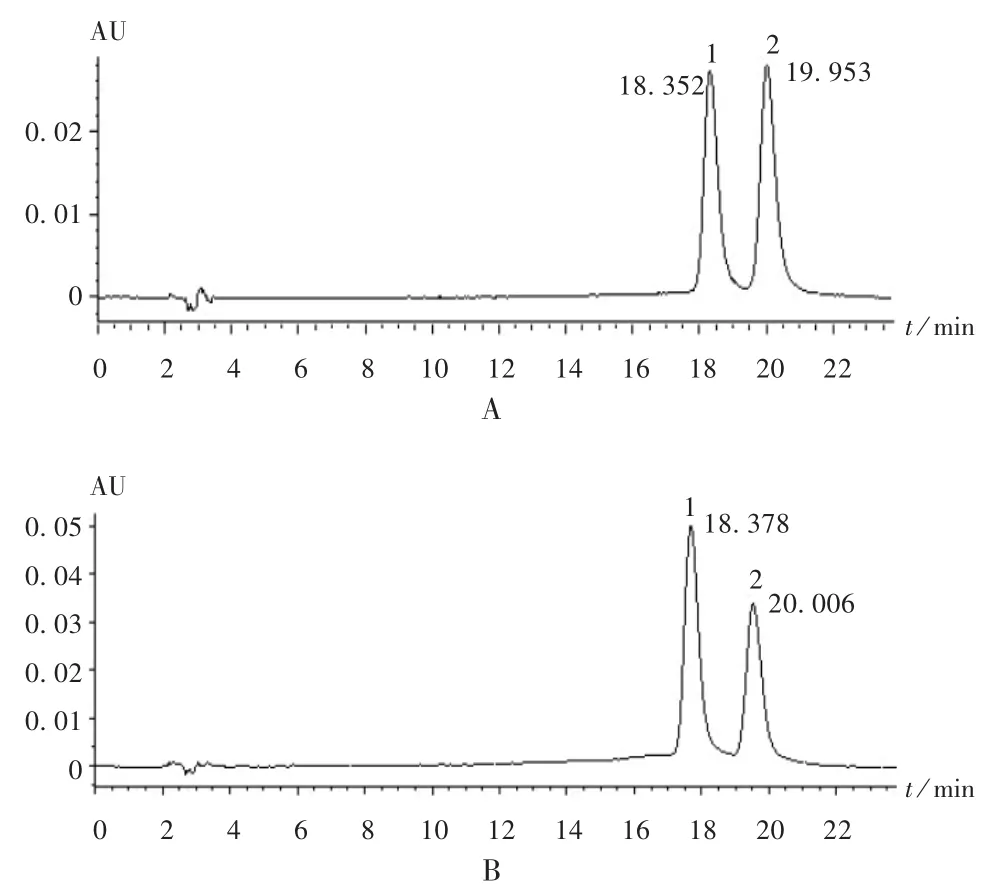
圖1 高效液相色譜圖
2.2 溶液制備
取姜黃素對照品和去甲氧基姜黃素對照品適量,精密稱定,用甲醇溶解,制成質量濃度姜黃素為20.04μg/mL、去甲氧基姜黃素為19.96 μg/mL的混合對照品溶液。取毛郁金根莖粉末1.0 g,精密稱定,置錐形瓶中,精密加入甲醇20 mL,密塞,稱定質量,超聲提取1 h,放至室溫,補足減失的質量,搖勻,過濾,取續濾液,再用0.45 μm微孔濾膜濾過,取續濾液作為供試品溶液。
2.3 方法學考察
標準曲線制備:精密吸取混合對照品溶液1,2,3,4,5,6 mL,分別置 10 mL容量瓶,加甲醇稀釋至刻度,搖勻,配成系列對照品溶液,以進樣量(X)為橫坐標、峰面積積分值(Y)為縱坐標進行線性回歸。結果見表1。
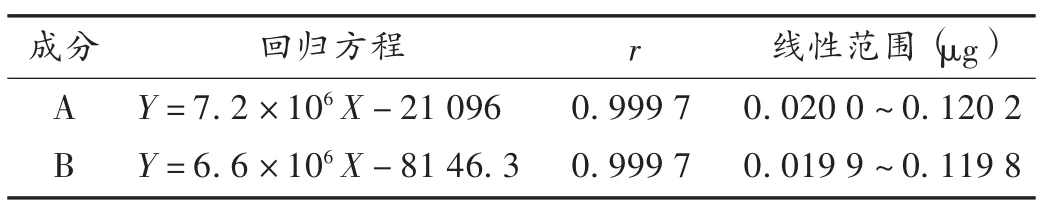
表1 毛郁金中2個姜黃素類成分回歸方程及線性范圍(n=6)
相對校正因子計算:取標準曲線制備項下制備的6個不同質量濃度的混合對照品溶液,分別進樣10 μL,以姜黃素為內標,在上述特定范圍內按照相對校正因子計算公式fis=fi/fs=(mi×As)/(ms×Ai)計算姜黃素對去甲氧基姜黃素的相對校正因子,其中i為某化合物,s為內標物,m為質量,A為峰面積。結果見表2。
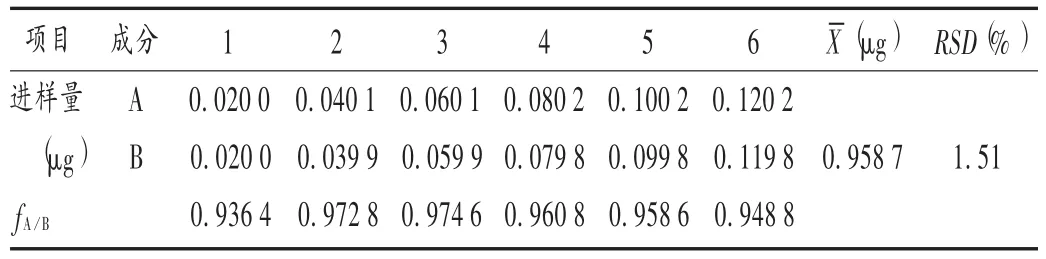
表2 不同濃度對照品溶液的相對校正因子(fA/B)
精密度試驗:取同一供試品溶液,精密吸取10 μL,連續進樣6次,測定峰面積。結果姜黃素和去甲氧基姜黃素峰面積日內精密度的RSD分別為1.50%和1.33%(n=6),表明儀器精密度良好。
穩定性試驗:取同一供試品溶液,分別于0,2,4,6,8,10 h時進樣,測定峰面積。結果姜黃素和去甲氧姜黃素面積的RSD分別為1.65%和1.38%(n=6),表明供試品溶液10 h內穩定性良好。
重復性試驗:取樣品粉末,依法制備供試品溶液6份,按色譜條件測定。結果姜黃素和去甲氧姜黃素的平均含量分別為0.111 3 mg/g和0.119 4 mg/g,RSD分別為1.76%和2.10%(n=6),表明方法重復性良好。
加樣回收試驗:取已知含量的毛郁金根莖粉末約0.5 g,共6份,精密稱定,置平底燒瓶中,分別加入精密稱定的姜黃素 60.12 μg,去甲氧姜黃素對照品 59.88 μg,依法制備供試品溶液,進樣測定。結果見表3。
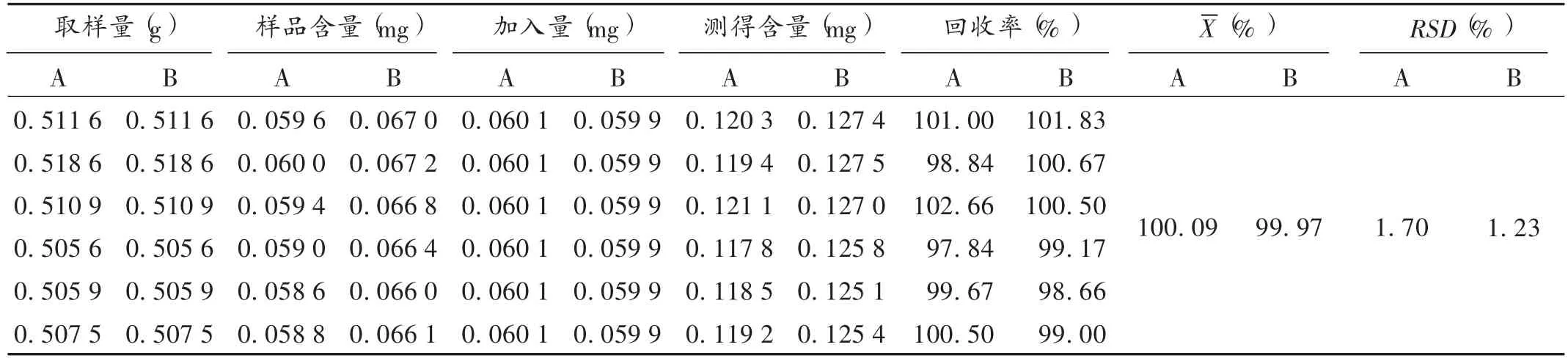
表3 加樣回收試驗結果(n=6)
2.4 校正因子的重復性考察
色譜柱考察:取標準曲線制備項下系列混合對照品溶液,分別進樣10 μL,以姜黃素為內標,計算姜黃素對去甲氧基姜黃素的校正因子,考察Hypersil BDS C18柱、Ultimate XB-C18柱和ECOSIL ODS-3柱的影響。結果見表4。可見,不同色譜柱所得相對校正因子相差不大。
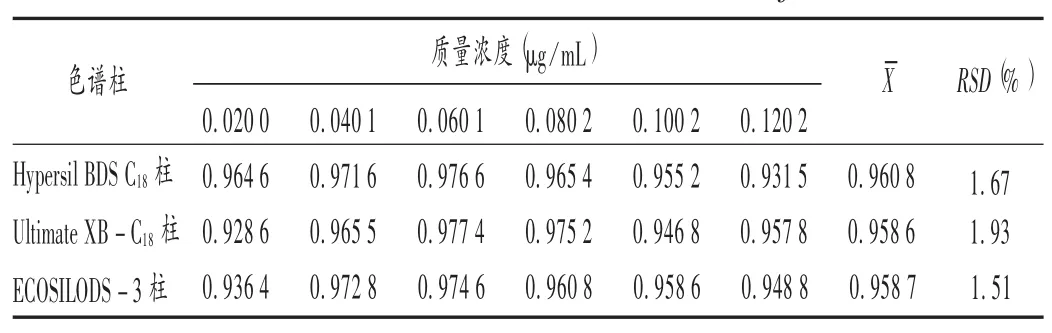
表4 不同色譜柱的相對校正因子(fA/B)
高效液相色譜考察:取上述混合對照品溶液,于Waters1525-1998型和威瑪龍UC3282-3292型高效液相色譜儀分別進樣各10 μL,以姜黃素為內標,計算姜黃素對去甲氧基姜黃素的校正因子,結果見表5,表明在不同液相色譜系統下所得的相對校正因子基本一致。

表5 不同高效液相色譜的相對校正因子
色譜峰專屬性考察:利用相對保留值(r=tR1/tR2)定位,即以待測組分與姜黃素色譜峰的相對保留時間來確定。去甲氧基姜黃素與姜黃素峰的相對保留時間的平均值為1.111 7,RSD為1.54% (n=6)。結果見表6。

表6 2種成分的保留時間及相對保留時間
2.5 一測多評法與外標法結果比較
取不同產地樣品粉末各1.0 g,精密稱定,依法制備供試品溶液,按擬訂色譜條件分別采用一測多評法和外標法計算含量。結果見表7。
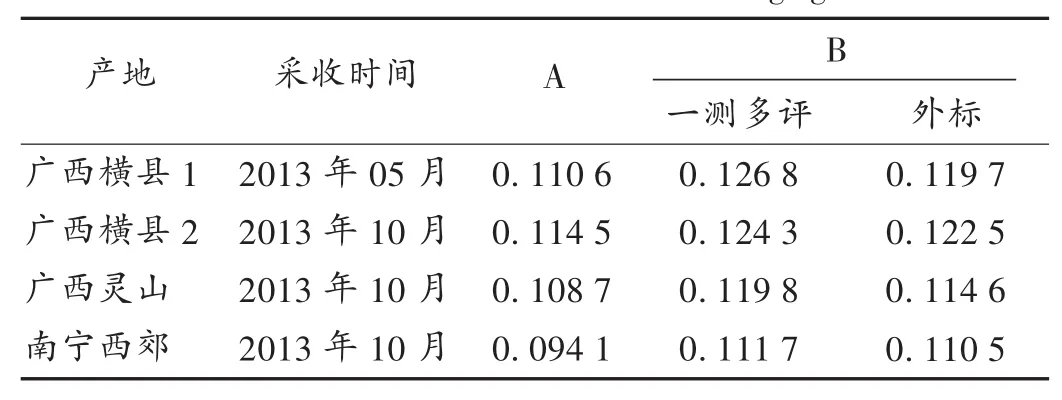
表7 不同產地毛郁金含量測定結果(mg/g,n=2)
3 討論
本研究中采用超聲、回流提取毛郁金藥材,結果顯示,兩者提取方式差異不明顯,而超聲提取方法簡單,故采用超聲提取。考察了甲醇、50%甲醇、50%乙醇作為毛郁金藥材的提取溶劑,結果表明用甲醇提取干擾雜質較少,噪音少,因此選用甲醇為提取溶劑。提取時間考察了30,60,90,120 min,結果顯示,超聲提取 60 min 基本可將供試品溶液中的姜黃素和去甲氧基姜黃素提取完全,90 min后其含量有所下降,故確定提取時間為60 min。
參照2015年版《中國藥典(一部)》莪術中姜黃素含量測定方法[11],以乙腈 -4%磷酸溶液(48∶52,V∶V)為流動相洗脫,姜黃素特征峰未能與去甲氧基姜黃素特征峰較好地分離。此方法對于毛郁金藥材姜黃素與去甲氧基姜黃素的分離效果不理想。通過反復試驗,選用乙腈-4%磷酸溶液(42∶58,V∶V)為流動相,色譜峰峰形尖銳,姜黃素特征峰與去甲氧基姜黃素特征峰能較好分離,樣品檢測25 min完成,節省了試驗成本和時間。
本研究首次采用“一測多評”法測定毛郁金藥材中姜黃素和去甲氧基姜黃素的含量,考察了不同品牌的色譜柱和色譜儀對姜黃素、去甲氧基姜黃素之間相對校正因子的影響,該方法與外標法測得的含量無顯著性差異,說明建立的不同化合物之間的校正因子具有較高的可信度,可在僅有一個對照品時通過一測多評技術實現同步測定藥材中多成分的指標成分,為毛郁金藥材的多指標質量控制模式提供了新方法。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
