壓力超負荷致小鼠心肌肥厚中miR-378對熱休克轉錄因子-1的調節作用
2019-09-23 12:27:22鄒云增
中國臨床醫學 2019年4期
苑 潔,鄒云增
復旦大學附屬中山醫院心內科,上海市心血管病研究所,上海 200032
心血管疾病是全球范圍內危害人類健康的“頭號殺手”。心肌肥厚是許多心臟疾病如高血壓、心瓣膜病、充血性心力衰竭及先天性心臟病的代償反應,會導致心力衰竭。而血流動力學壓力超負荷是引起心肌肥厚進而發生心力衰竭的主要因素。心臟在壓力負荷初期通常表現為代償性心肌肥厚,以滿足機體的各種生命活動的需要。然而當心臟在持續壓力超負荷情況下,代償性心肌肥厚會發展為失代償性心肌肥厚,從而誘發心功能不全和心力衰竭,增加猝死發生風險[1-2]。
熱休克蛋白轉錄因子1(HSF1)在許多病理條件的(缺血損傷、氧化應激、壓力超負荷等)刺激下均可以對心臟起保護作用,并在適應性的生理性心肌肥厚中發揮重要作用[1-5]。壓力超負荷下,HSF1能促進心臟血管新生、抑制心肌纖維化、維持心肌重構的早期代償[6-7],然而有關其調控機制的研究報道甚少。因此,研究HSF1在心肌肥厚中的調節機制,對于臨床治療中心功能的改善具有重要意義。
MicroRNA(簡稱miRNA)是廣泛存在于真核生物中長度約為21 nt的內源性非編碼RNA,參與基因的轉錄后調節,在生物體內的生理及病理過程中都扮演著重要角色[8-10]。已有研究證實,很多miRNAs參與心肌肥厚發生發展過程中重要基因的調控[9,11-12]。MiRNA是否也參與心臟保護因子HSF1的調控,值得進一步研究。
1 材料與方法
1.1 小鼠壓力超負荷模型(TAC)的建立 C57B/L6雄性小鼠,周齡為10~12周,體質量18~22 g,購于上海中科院斯萊克實驗動物有限責任公司。小鼠麻醉后臥于鼠板固定,氣管插管后連接呼吸機,沿胸骨左側的第二肋骨剪斷肋骨,在主動脈凸側分支的第一和第二分支之間將絲線穿過主動脈弓,用31G針頭造成升主動脈環行縮窄,之后逐層縫合。假手術組只穿線不結扎。
1.2 小鼠心臟超聲和血流動力學測定 對小鼠假手術組和TAC模型術后2周進行心功能和動脈收縮壓(ASP)測定。使用加拿大VisualSonics公司Vevo 770超聲診斷儀和30 MHz高頻探頭進行小鼠的心臟超聲檢測心臟功能,記錄B-Mode圖像,測量左室舒張期前壁厚度(LVAWd)、左室射血分數(LVEF)等指標。使用Millar導管對小鼠ASP進行測定。
1.3 心肌細胞培養 取1~3 d SD大鼠乳鼠15只,購于上海中科院斯萊克實驗動物有限責任公司。用75%乙醇消毒,取心尖大部用PBS洗2次,將組織剪碎。用0.1%胰酶溶液37℃消化5~6次,每次8 min,收集上清到DMEM標準培養基中終止消化,1 200r/min 離心5 min,將細胞沉淀收集到1個裝有DMEM低糖標準培養基中的收集管中,將收集得到的細胞懸液加入2個10 mm培養皿中,置于CO2培養箱進行差速貼壁法分離。2 h后,吸取上清,按照1×106/mL將細胞接種,次日換液,加入新鮮的DMEM低糖培養基。
1.4 MiR-378模擬物或抑制物細胞轉染 心肌細胞貼壁24 h后,換成無血清無抗生素低糖培養基預處理過夜,使用轉染試劑siPORTTMNeoFXTMTransfection Agent(Fisher, AM4510)轉染miR-378 模擬物 (35 nmol/L) (Applied Biosystems,AM17100)或抑制物(50 nmol/L) (Applied Biosystems,AM17000)分子以及FAM標記模擬物和抑制物的陰性對照。48 h后收集細胞或對細胞進行機械牽張刺激。
1.5 熒光素酶報告基因檢測 將擴增得到的HSF1野生型基因3′UTR或突變型片段各自插入到pMIR-REPORTTMluciferase miRNA expression(Applied Biosystems,AM4510) 報告載體中得到Luc-HSF1及種子序列突變載體Luc-HSF1-MU,使用FuGENE HD(Roche,E2311)根據分組將熒光素酶報告載體和miR-378模擬物或抑制物共轉染Cos7細胞,48 h后使用Dual-Light○RSystem(Applied Biosystems,T1003)檢測化學發光值。
1.6 miRNA表達水平檢測 用TRIzol試劑(InvitrogenTM,15596018)從心肌組織和心肌細胞中提取總RNA,并通過NanoDrop ND-1000分光光度計測量濃度。使用TaqMan MicroRNA Assays (Applied Biosystems,4427975)在ABI 7500系統進行qRT-PCR反應,U6作為內參,實驗步驟按照試劑盒操作。
1.7 Western 印跡檢測蛋白表達 心肌組織或心肌細胞用RIPA(碧云天,P0013C)裂解,混勻后4℃振蕩4 h,以12 000×g離心15 min(4℃),上清為總蛋白質。Western印跡按常規步驟電泳,每孔加入20~40 μg蛋白量,使用HSF1(CST,4356)、Hsp27(CST,95357)抗體和HRP偶聯的兔二抗(CST,7075)檢測各組蛋白表達水平,以GAPDH為內參計算其相對表達水平。

2 結 果
2.1 小鼠心肌組織內HSF1和miR-378的表達變化 小鼠心肌肥厚(圖1A)代償期時心肌HSF1表達顯著升高,而miR-378的表達顯著下降。血流動力學檢測結果顯示:TAC術后小鼠的ASP、收縮壓、左心室舒張末期壓力、收縮末期壓力均明顯升高(P<0.05),心肌肥厚處于代償期,心功能無顯著下降,LVEF值與假手術組相比無顯著差異(圖1B)。小鼠心臟超聲數據結果顯示:TAC手術2周后心肌肥厚顯著,心臟質量/體質量比值HW/BW、左室壁厚度顯著增加(P<0.05,圖1C)。TAC小鼠手術2周后心肌內HSF1及其下游調控蛋白Hsp27的表達均顯著升高(圖1D),而心肌內miR-378的表達顯著降低(圖1E)。

圖1 壓力超負荷導致心肌肥厚小鼠心肌內HSF1和miR-378的表達變化
A: 小鼠TAC 術后2周及假手術組心臟外觀、血流動力學變化;B:小鼠TAC術后ASP和LVEF表達變化; C:小鼠TAC術后LVAWd和HW/BW表達變化;D:Western印跡檢測TAC 術后2周小鼠心肌內HSF1和Hsp27蛋白表達變化; E: 實時定量PCR檢測TAC術后2周小鼠心肌內miR-378的表達變化.*P<0.05與假手術組相比;n=5
2.2 心肌細胞體外牽張刺激下HSF1和miR-378的表達變化 使用0.01%鼠尾膠原溶液按照6~10 μg/cm2的濃度包被硅膠皿,室溫下在無菌操作臺內將硅膠皿風干后。心肌細胞接種到硅膠皿培養后,將硅膠皿放在機械牽張板上拉伸,使得其長度可以增加20%。對體外培養的心肌細胞機械牽張刺激12 h、24 h,檢測心肌細胞內HSF1和miR-378的表達變化,發現HSF1的蛋白表達水平在牽張24 h后顯著升高(P<0.05,圖2A),miR-378的表達水平在機械牽張24 h后卻顯著下降(P<0.05,圖2B)。

圖2 心肌細胞體外牽張刺激下HSF1和miR-378的表達變化
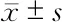
2.3 MiR-378對 HSF1的轉錄后調控 心肌細胞分別轉染miR-378模擬物(Pre-miR-378)、miR-378抑制物(Anti-miR-378)及FAM標記的陰性對照組(mock),24 h后分別在普通光鏡下和熒光顯微鏡下觀察細胞,發現90%以上的細胞發出綠色熒光,轉染效率達到90%(圖3A)。轉染48 h后,Real-time PCR結果顯示,轉染模擬物組心肌細胞中miR-378的表達量顯著升高,轉染抑制物組心肌細胞內源miR-378的表達被顯著抑制(圖3B)。檢測HSF1蛋白表達,轉染miR-378模擬物組HSF1的表達被顯著抑制(P<0.05),轉染miR-378抑制物組HSF1表達顯著升高(P<0.05,圖3C)。心肌細胞過表達miR-378能夠顯著抑制機械牽張引起的HSF1升高(圖3D)。
分析HSF1 3′UTR區域,發現在第1743~1765個堿基區域內有miR-378的潛在靶向結合種子序列(圖3E)。構建包含HSF1 3′UTR的熒光素酶報告基因載體Luc-HSF1及種子序列突變載體Luc-HSF1-MU,轉染Cos7細胞。當細胞過表達miR-378時,轉染Luc-HSF1組細胞內化學發光被顯著抑制,而Luc-HSF1-MU組則升高(P<0.05);當Cos7單獨轉染Luc-HSF1時,細胞內化學發光被顯著抑制,而當抑制內源miR-378時,化學發光的抑制作用被逆轉(P<0.05,圖3F)。因此,這些結果證實miR-378能夠通過靶向結合HSF1 3′UTR調控HSF1的表達。
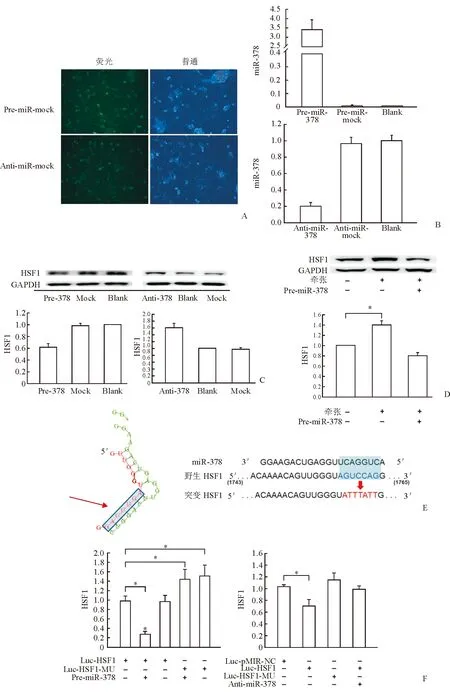
圖3 MiR-378對 HSF1的轉錄后調控
A:心肌細胞轉染FAM標記的miR-378模擬物和抑制劑(陰性對照),熒光普通光鏡觀察發現,轉染效率達90%; B:轉染miR-378模擬物和抑制物后,miR-378在心肌細胞中的表達情況; C:Western 印跡檢測心肌細胞過表達和抑制miR-378后HSF1的表達變化;D:Western 印跡檢測機械牽張刺激后心肌細胞內HSF1的蛋白表達;E:miR-378在HSF1 3′UTR的靶向結合位點,以及HSF1 3′UTR種子序列的突變型;F:熒光素酶保護基因檢測示miR-378可直接靶向結合HSF1 3′UTR.*P<0.05,n=3
3 討 論
HSF1在多種病理條件刺激下,如缺血損傷、氧化應激、壓力超負荷等,均對心臟起到一定的保護作用[3,6,13]。另外有研究證明,當機體內的HSF1表達降低時,心臟對外界刺激的適應性會隨之降低[14]。這些均表明在代償性心肌肥厚反應中,HSF1可能是一種生理性的保護因子,但是對于其引起這種保護性效應的機制,目前研究還是十分有限。
在心肌損傷中,一些miRNA與HSF1之間的調節作用被報道。HSF1能夠激活miR-135b的表達,促進內皮細胞的增殖[15];在心肌肥厚中,HSF1通過調節HSP70的表達促進miR-23表達的升高[16];在急性心梗的小鼠中,敲除內源性HSF1會促進miR-208的表達[17];房顫發生后,通過調節HSF1可引起miR-432表達的升高[18];miR-1可以上調HSF1的表達,當miR-1表達下調時會導致心肌肥厚的發生[19]。本研究通過構建心肌肥厚的代償模型,從在體和細胞水平同時觀察內源性HSF1和miR-378的表達變化,并對其中的調控機制進行了探討。
本研究中,TAC處理2周后,小鼠表現為代償期的病理性心肌肥厚,伴隨心肌內源性HSF1及其下游調控蛋白Hsp27的代償性升高,而心肌內源性miR-378的表達卻明顯下降。細胞水平發現機械牽張刺激心肌細胞會導致內源性miR-378的表達下降,而HSF1的表達則會升高。之前的研究[1-5]認為HSF1在適應性心肌肥厚中發揮重要作用,在心肌肥厚的早期代償階段表現為代償性的表達升高。那么在心肌組織中,HSF1的代償性升高是否受到miR-378的調控?本研究在心肌細胞中分別過表達和抑制miR-378,發現細胞過表達miR-378時會抑制HSF1的表達,而抑制miR-378的表達會使內源性HSF1的表達升高,并且在機械牽張刺激下,過表達miR-378會抑制HSF1的升高,證實了miR-378參與了HSF1的表達調控。利用生物信息學軟件miRanda和RNAhybrid分析miR-378的潛在靶點,發現HSF1的3′UTR上有miR-378的靶向結合位點,且最小自由能為-28.4 kal/mol,說明其可以為miR-378的結合提供很好的空間結構,本研究進一步利用熒光素酶報告基因證實了miR-378可以靶向結合HSF1 3′UTR,進而調控其轉錄后的翻譯。
綜上所述,HSF1作為心肌內源重要的保護因子,在心肌缺血缺氧、機械應力及炎癥損傷中均發揮了重要的保護作用。在高血壓心肌肥厚的早期代償階段,心肌miR-378表達的下降使得其對內源性HSF1轉錄后抑制作用減弱,進而對HSF1的代償性升高發揮了重要的調控作用。由于miRNA表達及調控的時序性的復雜性,有關miR-378通過調節HSF1在心肌肥厚發生發展階段所發揮的生物學功能需要進一步深入研究,同時miR-378與miR-1是否協同調節HSF1也需要深入探討。該研究對于HSF1參與調節心肌保護的機制提供了新的理論基礎,為心肌重構干預靶點的開發提供了新思路。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
海峽科技與產業(2016年3期)2016-05-17 04:32:12
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
