蛋白激酶D1對心肌梗死大鼠心肌組織炎癥和凋亡的影響
2019-09-24 08:11:40毛秉豫
中國藥理學通報 2019年10期
關鍵詞:模型
楊 雷,劉 萍,劉 暖,王 倩,毛秉豫
(南陽理工學院河南省張仲景方藥與免疫調節重點實驗室,河南 南陽 473004)
2017年,心血管病研究報告指出,2015年中國城市、農村心血管病死亡占比分別高達42.61%和45.01%,心肌梗死是引發死亡的主要原因之一[1]。心梗后梗死區域周邊和非梗死區心肌組織容易發生細胞凋亡,進而改變心肌的血流動力學參數,造成心臟收縮和舒張功能障礙,引發心室重塑[2]。因此,預防和控制心肌細胞凋亡是減緩心室重塑的有效對策之一[2]。
心梗心肌組織缺血損傷后,炎癥反應調控蛋白如Toll樣受體4(Toll-like receptor 4,TLR4)、黏膠蛋白/肌腱蛋白C(Tenascin-C,TN-C)、核轉錄因子κB(nuclear factor kappa B,NF-κB)p50、NF-κB p65,炎癥介質如白細胞介素1(interleukin-1,IL-1)、IL-6等,以及凋亡調控蛋白Bcl-2、caspase-3、Bax激活,啟動炎癥和凋亡反應,進而引發心室的不良重構[3]。蛋白激酶D1(protein kinase D1,PKD1)在心血管系統中,參與血管新生、心肌收縮、心肌重塑等進程[4-6],但其對心肌梗死后心肌細胞凋亡和炎癥反應的調控作用尚不清楚。本研究旨從分子生化角度,分析PKD1給藥對大鼠心肌梗死后炎癥和凋亡的影響。
1 材料
1.1 實驗動物45只Wistar大鼠,♂,SPF級,8周齡,體質量(200±20)g,購自北京維通利華實驗動物技術有限公司,動物生產許可證號:SCXK(京)2016-0011。實驗在南陽理工實驗動物倫理委員會(批準號:NYIST-20180128)指導下進行。
1.2 藥物與試劑PKD1,購自美國Pierce公司;caspase-3、Bax、Bcl-2一抗,購自美國Sigma公司;TLR4、TN-C、NF-κB p50、NF-κB p65一抗及RNA提取試劑盒,均購自Santa Cruz上海公司;TUNEL染色試劑盒,購自北京百奧萊博公司;RNA PCR Kit (AMV) Ver.3.0試劑盒,購自大連寶生物公司;IL-1、IL-6、NF-κB p50和NF-κB p65的TaqMan探針,由Annoron北京公司代理設計。
1.3 儀器CUT6062型病理切片機(德國SLEE公司); NanoDrop-ND2000型紫外超微分光光度計(美國Thermo Fisher公司);T10高速組織勻漿機(德國IKA公司);Nikon Tis型熒光顯微鏡及配套分析軟件NIS-Elements Software BR(日本尼康公司)。
2 方法
2.1 動物造模和分組將大鼠隨機分為假手術(Sham)組、模型(Model)組和PKD1組,每組15只。模型組和PKD1組參照之前文獻的方法[5],通過永久性結扎左前降支冠狀動脈制作心肌梗死模型;Sham組僅手術而不結扎血管。術后,PKD1組給予1 mg·kg-1·d-1的PKD1,Sham和模型組給予等量生理鹽水,均灌胃7 d。在7 d結束時,30 mg·kg-1戊巴比妥鈉腹腔注射麻醉大鼠,進行血流動力學評估。評估結束,頸總動脈放血法處死動物,進行相關實驗分析。
2.2 血流動力學評估將SPR-838壓力容積導管經左頸動脈插入麻醉后大鼠的左心室(left ventricle, LV)腔內,接入壓力換能器,應用BL-420F生物機能實驗系統,記錄LV舒張末期容積(end-diastolic volume,EDV)、收縮末期容積(end-systolic volume,ESV)、等容舒張常數(isovolumic relaxation constant,Tau)、最大壓力導數(maximum derivative of pressure,Max dp/dt)、最小壓力導數(minimum derivative of pressure,Min dp/dt)等數據。
2.3 HE染色取處死后大鼠的心臟,分離左心室和室間隔前2/3,參照之前文獻的方法[5],4%多聚甲醛固定,包埋,制作4 μm厚石蠟切片,HE染色,光學顯微鏡×400放大視野下對心肌組織進行分析。
2.4 TUNEL染色先制作石蠟切片,再參照試劑盒說明書進行TUNEL染色,細胞核DAPI復染。熒光顯微鏡下正常細胞示藍色,陽性凋亡心肌細胞示紅色。每張玻片選5個非重復視野,計數陽性細胞個數。
2.5 免疫組化取材同HE染色,參照之前文獻的方法[5],將病理切片常規脫蠟、修復并消除內源性過氧化物酶,PBS液沖洗后,加入抗caspase-3、Bax、Bcl-2的一抗,置4 ℃孵育12 h后,PBS沖洗清除表面雜質和一抗;加入羊抗兔二抗,常溫孵育15 min后,加入DAB顯色液孵育3 min,顯微鏡下觀察,陽性細胞呈現黃色或者棕黃色。每張玻片選5個非重復視野,計數caspase-3、Bax、Bcl-2陽性細胞個數。
2.6 免疫印跡取心尖部心肌組織制成組織勻漿后,RIPA細胞裂解液裂解蛋白,應用經典的BCA法測定濃度并制作標準曲線。取30 μg總蛋白,94 ℃變性3 min后,進行12% SDS-PAGE電泳,轉移到PVDF膜,用含5%脫脂乳的TBS緩沖液封閉1 h,添加caspase-3、Bax、Bcl-2、TLR4、TN-C、NF-κB p50、NF-κB p65等一抗,4 ℃孵育12 h。PBS液沖洗3次,二抗(1∶2 000稀釋)孵育1 h后,TBST洗膜,暗室曝光顯影。計算并記錄各樣本蛋白與β-actin相對灰度值。
2.7 實時定量PCR(qPCR)檢測根據RNA提取試劑盒操作說明,提取心肌組織總RNA,NanoDrop-ND2000紫外超微分光光度計基于260 nm/280 nm比值監控質量,取1 μL RNA,用RNA PCR Kit(AMV) Ver.3.0試劑盒逆轉錄為cDNA。IL-1、IL-6、NF-κB p50和NF-κB p65基因的mRNA表達采用TaqMan市售水解探針檢測,PCR擴增條件:94 ℃,2 min(預變性);95 ℃,30 s(變性);65 ℃,30 s(退火);60 ℃,30 s(延伸);40個循環。記錄各個反應的Ct值,以β-actin基因為內參,ΔΔCt法分析結果,按照公式2-ΔΔCt計算各基因相對于β-actin內參基因的表達水平。引物序列見Tab 1。

3 結果
3.1 PKD1對心梗大鼠血流動力學的影響Tab 2結果顯示,與Sham組相比,模型組大鼠EDV和ESV值明顯升高(P<0.01),而Max dp/dt和Min dp/dt值明顯降低(P<0.01);與模型組相比,PKD1治療組大鼠EDV和ESV值明顯降低(P<0.01),而Max dp/dt和Min dp/dt值明顯升高(P<0.01);PKD1組和模型組Tau值均明顯低于Sham組,但兩組間無統計學差異。提示PKD1可改善EDV、ESV、Max dp/dt、Min dp/dt等血流動力學參數,對Tau沒有影響。
3.2 PKD1對心梗大鼠組織形態學的影響Fig 1的HE染色結果表明,Sham組大鼠組織結構規則、紅色心肌組織清晰,未見炎性細胞浸潤;模型組大鼠心肌組織壞死比例較高,炎性細胞浸潤和成纖維細胞增生明顯;PKD1治療組大鼠心肌組織恢復較好,細胞核較為清晰,少見炎性細胞浸潤,但細胞核仍顯肥大。

Tab 1 Sequence of PCR primer

Tab 2 Effect of PKD1 on hemodynamics in rats with myocardial infarction n=15)
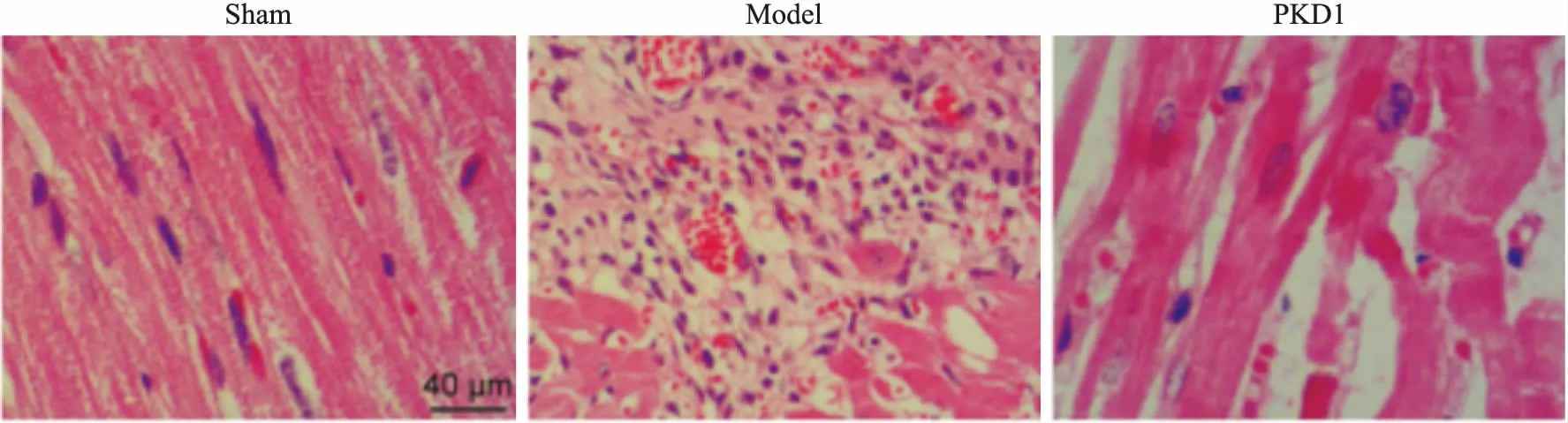
Fig 1 Effects of PKD1 on histomorphology of myocardial infarction rats(HE, ×400)
3.3 PKD1對心梗大鼠心肌細胞凋亡的影響Fig 2的TUNEL染色分析表明,Sham組和PKD1組大鼠心肌細胞主要發出藍色熒光,個別細胞發出紅色熒光,模型組大鼠心肌細胞主要呈紅色熒光。細胞計數結果表明,Sham組和PKD1組大鼠呈紅色熒光的陽性凋亡細胞個數均明顯少于模型組(P<0.01)。免疫組化和Western blot結果均表明(Fig 2、3),模型組大鼠心肌組織中caspase-3、Bax表達明顯高于Sham組,而Bcl-2表達明顯低于Sham組(P<0.01);PKD1治療后,大鼠心肌組織中caspase-3、Bax表達較模型組明顯降低(P<0.01),而Bcl-2表達明顯升高(P<0.01)。提示PKD1治療可有效對抗心梗大鼠心肌組織受損后引起的心肌細胞凋亡。
3.4 PKD1對心梗大鼠心肌組織中炎癥反應調控蛋白表達的影響Fig 4的結果證實,PKD1干預可明顯下調模型組大鼠因心肌缺血損傷引起的TLR4、TN-C、NF-κB p50和NF-κB p65的表達升高(P<0.01)。表明PKD1給藥可有效抑制心梗大鼠心肌組織中的炎癥反應。
3.5 PKD1對心梗大鼠心肌組織中炎癥因子mRNA表達的影響如Fig 5所示,PKD1干預可明顯下調模型組大鼠因損傷所致的致炎因子IL-1、NF-κB p50和NF-κB p65的mRNA表達升高(P<0.01),但PKD1組和模型組間IL-6 mRNA表達沒有統計學差異。表明PKD1可下調IL-1、NF-κB p50、NF-κB p65 mRNA表達,但不影響IL-6 mRNA表達。
4 討論
本研究利用大鼠心肌梗死模型,評價PKD1在心肌梗死心肌組織損傷中的抗炎、抗凋亡作用。研究發現,PKD1治療1周可有助于減輕心肌梗死后缺血心肌組織中的炎癥和凋亡反應。
ESV和Max dp/dt反映心臟的收縮功能,EDV和Min dp/dt反映心臟的舒張功能。PKD1可明顯降低ESV和EDV的值,同時上調Max dp/dt和Min dp/dt的值。表明PKD1干預可改善心肌的順應性,提高心功能指標,在改善心梗大鼠心肌組織的血流動力學方面效果明顯。HE染色結果表明,心肌梗死后組織壞死和炎性細胞浸潤明顯,PKD1治療可明顯減輕心肌組織的壞死程度,但心肌細胞仍明顯肥大,這可能與用藥時間較短有關,也可能是PKD1并不明顯抑制心肌肥大。
TUNEL染色結果表明,心肌梗死后出現大量凋亡的心肌細胞,而PKD1治療可有效抑制心肌細胞的凋亡。免疫組化和免疫印跡結果均表明,心肌梗死后,心肌組織內caspase-3、Bax的表達明顯升高,Bcl-2的表達明顯降低,而PKD1治療可有效抑制caspase-3、Bax的上調,并促進Bcl-2的表達上調。文獻研究表明[7-8],caspase-3、Bax的表達增多,而Bcl-2表達減少意味著組織損傷的加重和細胞凋亡數量的增多;反之,則表明損傷的減輕和凋亡細胞數量的減少。從本實驗結果分析,PKD1治療能有效抑制心梗后心肌細胞的凋亡。
心肌梗死后引發缺血損傷的起始階段,很多炎癥因子被損傷刺激激活,趨向至損傷區域[9]。在這一階段,炎癥白細胞,如單核細胞、巨噬細胞和中性粒細胞數量激增。TLRs是心肌缺血損傷后白細胞表達的模式識別受體[10],也是一種重要的炎癥反應調控蛋白,可調控NF-κB的轉運,或者影響炎性細胞因子如IL-1、IL-6、TNF-α等的釋放反應[11]。同時,損傷反應也會刺激誘導炎癥白細胞釋放更多的炎癥細胞因子,激發規模更大的炎癥反應,致使梗死范圍進一步擴大[12]。其中,IL-1不僅可導致心肌梗死后炎癥反應加重,還可引發心肌細胞凋亡、纖維化、心肌肥厚等不良后果[13]。心肌細胞凋亡又會進一步導致補體系統和TLRs的激活,并生成大量的氧自由基,進而激活心肌細胞核因子NF-κB,誘發趨化因子、細胞因子或黏附分子生成增多[14]。缺氧、活性氧以及TNF-α、IL-1介導的炎性刺激,均可激活NF-κB[14]。在細胞核中,NF-κB p50/RelA及NF-κB p65可誘導激發IL-1、IL-6、TNF-α、急性期反應蛋白以及黏附分子ICAM-1和YCAM1等的生成增多,參與炎癥反應的進程[14]。而且心肌梗死小鼠NF-κB p50基因敲除后,心室重塑可以被逆轉[14]。因此,就本研究結果來看,PKD1治療的有益作用可能與其下調TLR4、IL-1、NF-κB p50和NF-κB p65的表達密切相關,進而逆轉心室重塑和改善心肌功能。本研究還發現,PKD1干預不改變IL-6的表達水平,這可能是PKD1對炎性細胞因子的作用具有一定的選擇性。
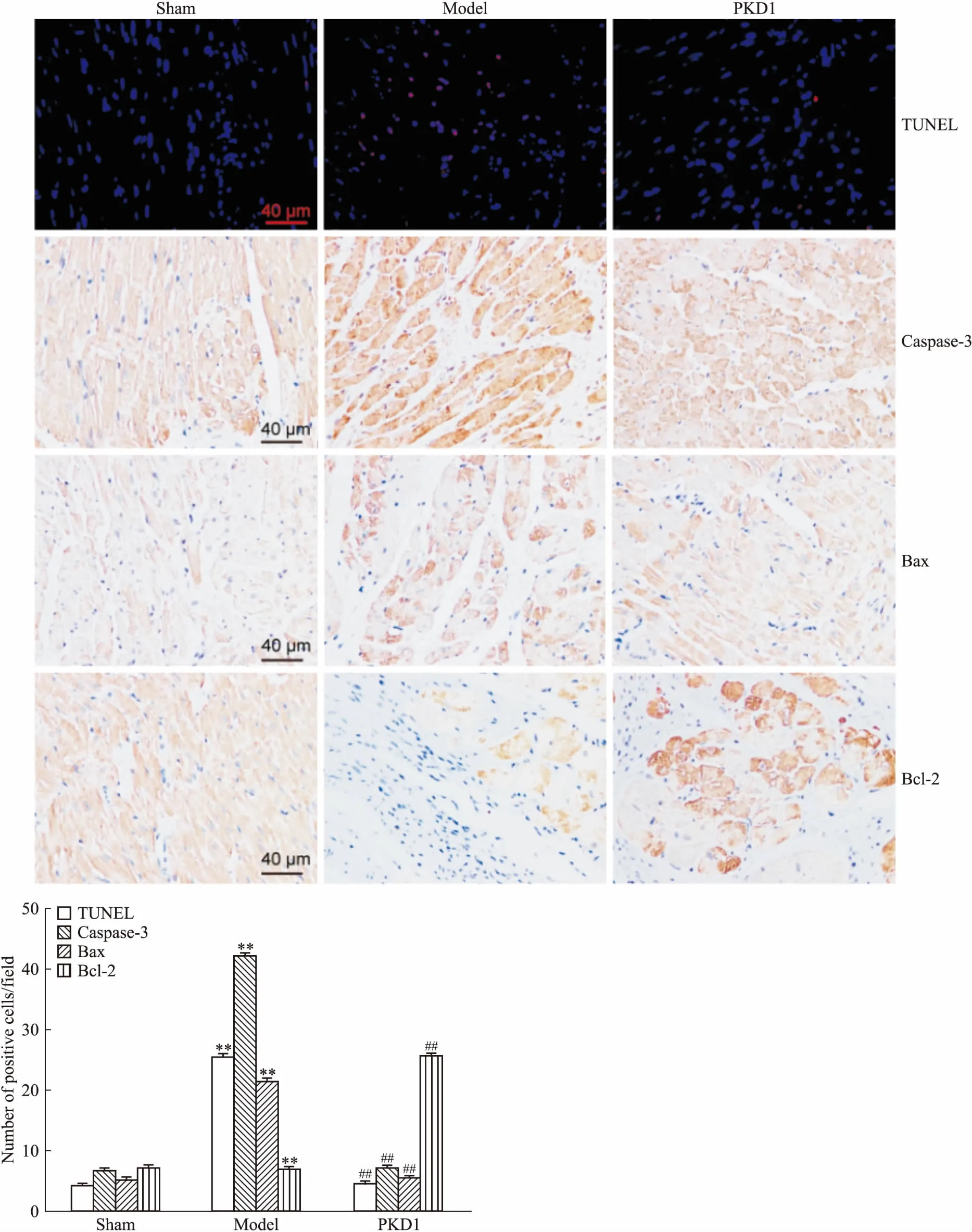
Fig 2 Effect of PKD1 on apoptosis of myocardial cells in rats with myocardial infarction vs sham group;##P<0.01 vs model group
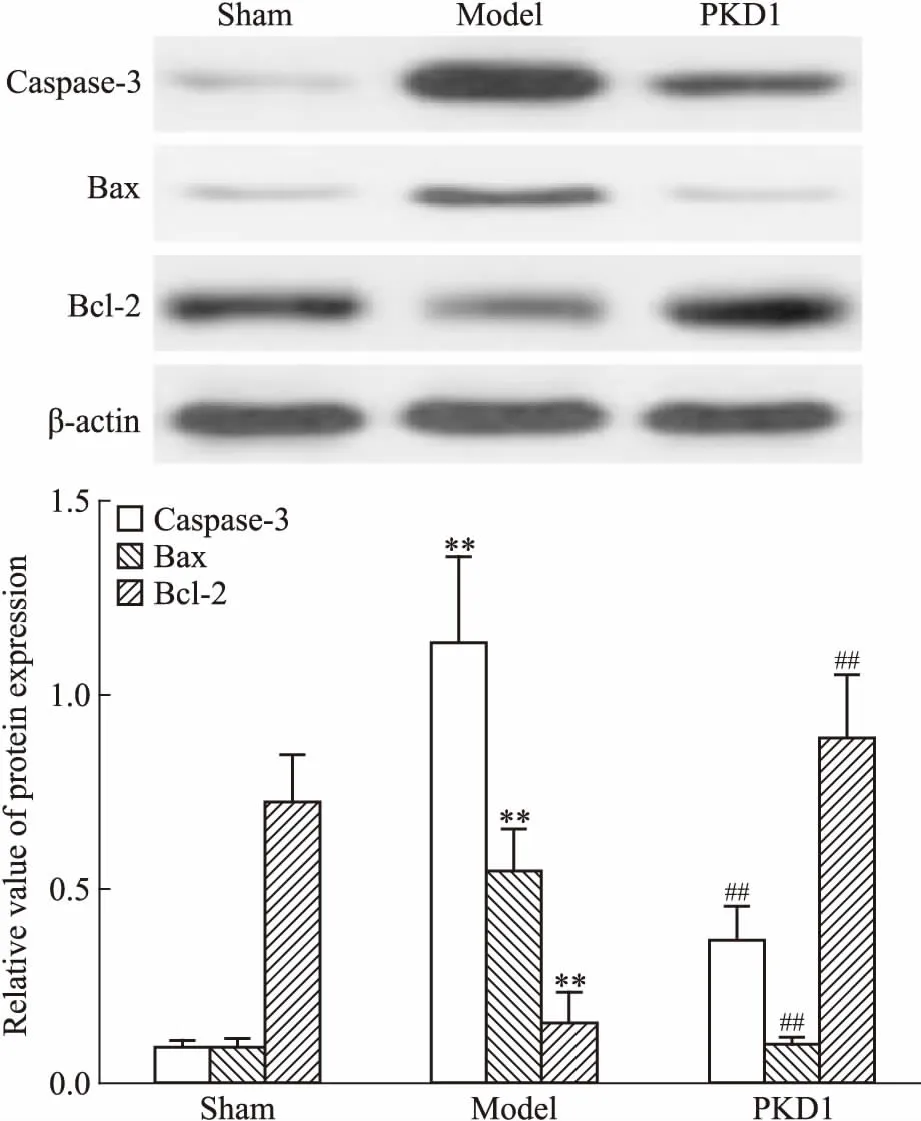
Fig 3 Effect of PKD1 on expression of apoptotic regulatory protein in myocardial tissue of rats with myocardial infarction

Fig 4 Effect of PKD1 on expression of inflammatory regulatory protein in myocardial tissue of rats with myocardial infarction

Fig 5 Effects of PKD1 on mRNA expression of inflammatory factors in myocardium of rats with myocardial infarction
TN-C也屬于炎癥的重要標志物之一,其本身屬于一種基質細胞蛋白,參與新膠原分子的形成,并具有調節NF-κB活化的作用[15]。敲除TN-C基因可下調心肌梗塞后心肌組織膠原纖維的占比,增強心肌的順應性,抑制心衰的發生[14]。本實驗發現,PKD1治療可降低TN-C的表達水平,這可能是通過下調NF-κB p50和NF-κB p65表達而實現的。
以上研究表明,PKD1具有調節心肌梗死大鼠心肌組織的炎癥反應,減輕梗死后心肌細胞凋亡,改善梗死后血流動力學參數,進而逆轉心室重塑的潛在作用。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19