異甘草酸鎂注射液與注射用環磷腺苷葡胺配伍的穩定性*
2019-10-11 02:08:20趙雯雯豆興茹黃春燕陳明圓李軼
醫藥導報 2019年10期
關鍵詞:標準
趙雯雯,豆興茹,黃春燕,陳明圓,李軼
(蘇州大學附屬第一醫院藥學部,蘇州 215000)
異甘草酸鎂注射液為臨床常用的甘草酸制劑,是用于抗炎保肝的一線藥物[1]。注射用環磷腺苷葡胺是環磷腺苷與葡甲胺(摩爾比為1:1)的鹽,是一種非洋地黃類強心劑。有研究發現,注射使用環磷腺苷能直接補充血漿和肝細胞內cAMP含量,促進肝細胞內ATP合成,還能抑制炎癥細胞的粘附和浸潤,從而減輕肝臟的炎癥反應、恢復肝功能[2]。
臨床上常選擇異甘草酸鎂注射液與注射用環磷腺苷葡胺配伍的方案用于某些特殊的情況,說明書未提及二者之間是否存在配伍禁忌,也未檢索到兩種藥品在不同輸液中配伍的報道。因此,筆者將異甘草酸鎂注射液與注射用環磷腺苷葡胺配伍溶于5%葡萄糖注射液、10%葡萄糖注射液、0.9%氯化鈉注射液、5%葡萄糖氯化鈉注射液、轉化糖注射液,考察其配伍后穩定性,為患者的用藥安全提供初步的理論支持,報道如下。
1 儀器與試藥
1.1儀器 Agilent Technologies 1100型高效液相色譜儀(美國安捷倫公司),包括G1314A可變波長掃描紫外檢測器、G1316A柱溫箱、G1379A脫氣機、G1311A四元泵、G1313A自動液體進樣器,Agilent ChemStation for LC and LC/MS systems數據分析系統;電子分析天平(METTLER TOLEDO公司,型號:XS105 DualRange,感量:0.01 mg);pH/ORP測試儀(上海羅素科技有限公司,型號:RPB1000);;GWJ-5微粒檢測儀 (天津天大天發科技有限公司)。
1.2藥品與試藥 異甘草酸鎂標準品(中國食品藥品檢定研究院,批號:100879-201302);環磷腺苷標準品(中國食品藥品檢定研究院,批號:140709-201404);異甘草酸鎂注射液(正大天晴藥業集團股份有限公司,批號:171222204);注射用環磷腺苷葡胺(無錫凱夫制藥有限公司,批號:17111164);乙腈(色譜純);磷酸(分析純)。
2 方法與結果
2.1色譜條件 色譜柱[依利特,填料:SinoChrom ODS-BP(4.6 mm×150 mm,5 μm)];流動相:1%磷酸-乙腈(55:45);流速:1 mL·min-1;檢測波長:250 nm;柱溫:25 ℃;進樣量:10 μL。
2.2溶液的制備
2.2.1標準品溶液 分別精密稱取異甘草酸鎂標準品11.02 mg和環磷腺苷標準品10.00 mg,分別置于10 mL量瓶,加純化水定容,搖勻制成濃度均為1.0 mg·mL-1的標準品儲備液。分別取異甘草酸鎂和環磷腺苷標準品儲備液適量,配制成濃度為0.4,0.5,0.6,0.7,0.8 mg·mL-1的異甘草酸鎂和環磷腺苷標準品溶液。
2.2.2供試品溶液 按照臨床配伍的濃度,將異甘草酸鎂注射液200 mg和注射用環磷腺苷葡胺180 mg溶于250 mL的5%葡萄糖注射液、10%葡萄糖注射液、0.9%氯化鈉注射液、5%葡萄糖氯化鈉注射液、轉化糖注射液,5種不同的溶媒配制成含有異甘草酸鎂0.69 mg·mL-1、注射用環磷腺苷葡胺0.62 mg·mL-1的混合溶液作為供試品溶液,考察2種藥物配伍在不同溶媒中24 h的穩定性。以此方法配制2份,第1份置于25 ℃常溫避光保存,作為供試品溶液一,第2份置于4 ℃保存,作為供試品溶液二。
2.3方法學驗證
2.3.1專屬性實驗 取0.6 mg·mL-1異甘草酸鎂標準品溶液、0.6 mg·mL-1環磷腺苷標準品溶液、5%葡萄糖注射液、10%葡萄糖注射液、0.9%氯化鈉注射液、5%葡萄糖氯化鈉注射液、轉化糖注射液按上述色譜條件依次進樣,每個樣品分析6 min,記錄色譜圖,見圖1。結果表明,在此色譜條件下,空白溶媒對異甘草酸鎂和環磷腺苷的含量測定并無較大影響,同時,兩者的峰對稱性良好,且互不干擾,其專屬性良好。
2.3.2線性關系考察 將濃度為0.4,0.5,0.6,0.8,1.0 mg·mL-1異甘草酸鎂和環磷腺苷標準品溶液,按上述色譜條件依次進樣,分析時間為6 min,以峰面積(Y)為縱坐標與濃度(X)繪制標準曲線,得到回歸方程:Y1=7 264.6X+5.972 4,r=0.999 8和Y2=18 639X+1 394.1,r=0.999 0。結果表明,異甘草酸鎂和環磷腺苷的濃度在0.4~1.0 mg·mL-1范圍內與峰面積線性關系良好。
2.3.3精密度實驗 取0.5 mg·mL-1異甘草酸鎂和環磷腺苷標準品溶液按上述色譜條件各連續進樣6次,每次分析6 min。記錄峰面積,計算峰面積相對標準偏差(RSD)值,異甘草酸鎂RSD為0.09%,環磷腺苷RSD為0.05%。連續3 d重復上述操作,記錄峰面積,計算RSD值,異甘草酸鎂為0.29%,環磷腺苷為0.45%,表明精密度良好。
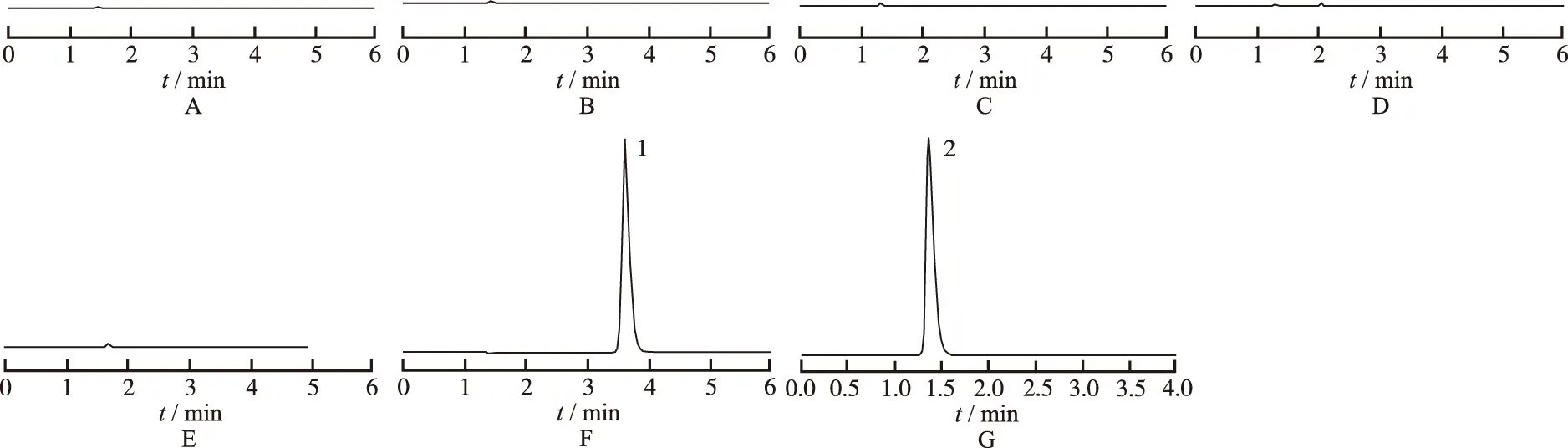
A.5%葡萄糖注射液;B.10%葡萄糖注射液;C.0.9%氯化鈉注射液;D.5%葡萄糖氯化鈉注射液;E.轉化糖注射液;F.異甘草酸鎂標準品;G.環磷腺苷標準品;1.異苷草酸鎂;2.環磷腺苷。
2.3.4穩定性實驗 取0.7 mg·mL-1異甘草酸鎂標準品兩份,分別在25和4 ℃保存條件下,于0,1,3,6,10,24 h依次進樣,測定峰面積,計算其RSD分別為0.31%和0.34%。取0.6 mg·mL-1環磷腺苷標準品兩份,同法測定峰面積,計算其RSD分別為0.54%和0.34%。表明兩種標準品在25和4 ℃保存條件下,24 h內基本穩定。
2.3.5加樣回收實驗 精密量取含異甘草酸鎂0.69 mg·mL-1、環磷腺苷葡胺0.62 mg·mL-1的5%葡萄糖注射液9份,每份0.5 mL,分別精密加入含異甘草酸鎂標準品0.276,0.345,0.414 mg的儲備液制成混合溶液,每個濃度平行配置3份,搖勻,取液按以上述色譜條件進樣,記錄異甘草酸鎂峰面積,根據標準曲線計算異甘草酸鎂含量和回收率,并計算平均回收率為102.25%。結果表明,該方法測定異甘草酸鎂含量的準確度良好。
同法精密加入含0.248,0.310,0.372 mg環磷腺苷標準品的儲備液,制成混合溶液按以上述色譜條件進樣,記錄異甘草酸鎂峰面積,根據標準曲線計算異甘草酸鎂含量和回收率,并計算回收率的平均值為102.47%。結果表明,該方法測定環磷腺苷含量的準確度良好。
2.4供試品外觀及pH值測定 按臨床常用量將“2.2”項配制好的供試品溶液5組,分別在25和4 ℃放置,加藥后于0,1,3,6,10,24 h取樣,觀察其外觀變化,測定pH值,結果見表1。結果發現,5組溶液于不同溫度下外觀在24 h內均呈現無色澄清透明,無沉淀產生。
2.5供試品含量測定 將“2.2”項配制好的供試品溶液于0,1,3,6,10,24 h取樣,進行異甘草酸鎂注射液和注射用環磷腺苷葡胺的含量測定。實驗結果見表2。在25和4 ℃條件下,24 h內含量變化在95%~105%,在臨床用藥的有效范圍內,24 h色譜圖無其他吸收峰產生。
2.6不溶性微粒的檢測 《中華人民共和國藥典》2015年版規定,標識裝量≥100 mL的靜脈用注射液,除另有規定外,每毫升中含粒徑≥10 μm微粒數不得超過25粒,含粒徑≥25 μm微粒數不得超過3粒[3]。
將上述“2.2”配制好供試品溶液5組,分別在25和4 ℃放置,加藥后0,1,3,6,10,24 h取樣,對供試品溶液的不溶性微粒進行檢測,實驗結果見表3,微粒變化不超過《中華人民共和國藥典》要求。

表1 不同溫度下24 h內pH值變化
3 討論
HPLC測定含量需要探索適合的色譜條件,筆者參考文獻[4-6],確定流動相為1%磷酸:乙腈(55:45),配伍溶液在進樣6 min內出現明顯環磷腺苷和異甘草酸鎂峰,兩者峰對稱性良好,且互不干擾,峰形良好。由于環磷腺苷葡胺中葡甲胺在反相色譜中幾乎不保留,故將環磷腺苷作為含量測定對象[7-8]。含量測定的方法學研究主要考察了專屬性、精密度和線性關系,考察指標均顯示良好,該方法具有較高的可行性。
本次研究考察臨床常用異甘草酸鎂注射液和注射用環磷腺苷葡胺配伍溶于5種不同溶媒分別在25和4 ℃下放置,24 h內外觀、pH值、含量以及不溶性微粒的變化,發現配伍溶液在24 h外觀均呈現無色澄清透明,無沉淀產生,不溶性微粒符合要求。溶液配置24 h內,除以0.9%氯化鈉注射液為溶媒的配伍溶液pH值約為6.0,其他配伍溶液pH值均保持在5.2~5.6。5種溶酶中異甘草酸鎂和環磷腺苷含量變化均在(100±5)%。實驗結果表明,異甘草酸鎂200 mg和注射用環磷腺苷葡胺180 mg配伍溶于250 mL的5%葡萄糖注射液、10%葡萄糖注射液、0.9%氯化鈉注射液、5%葡萄糖氯化鈉注射液、轉化糖注射液,24 h內穩定性良好,可配伍使用,從而為臨床用藥提供了初步的理論依據。
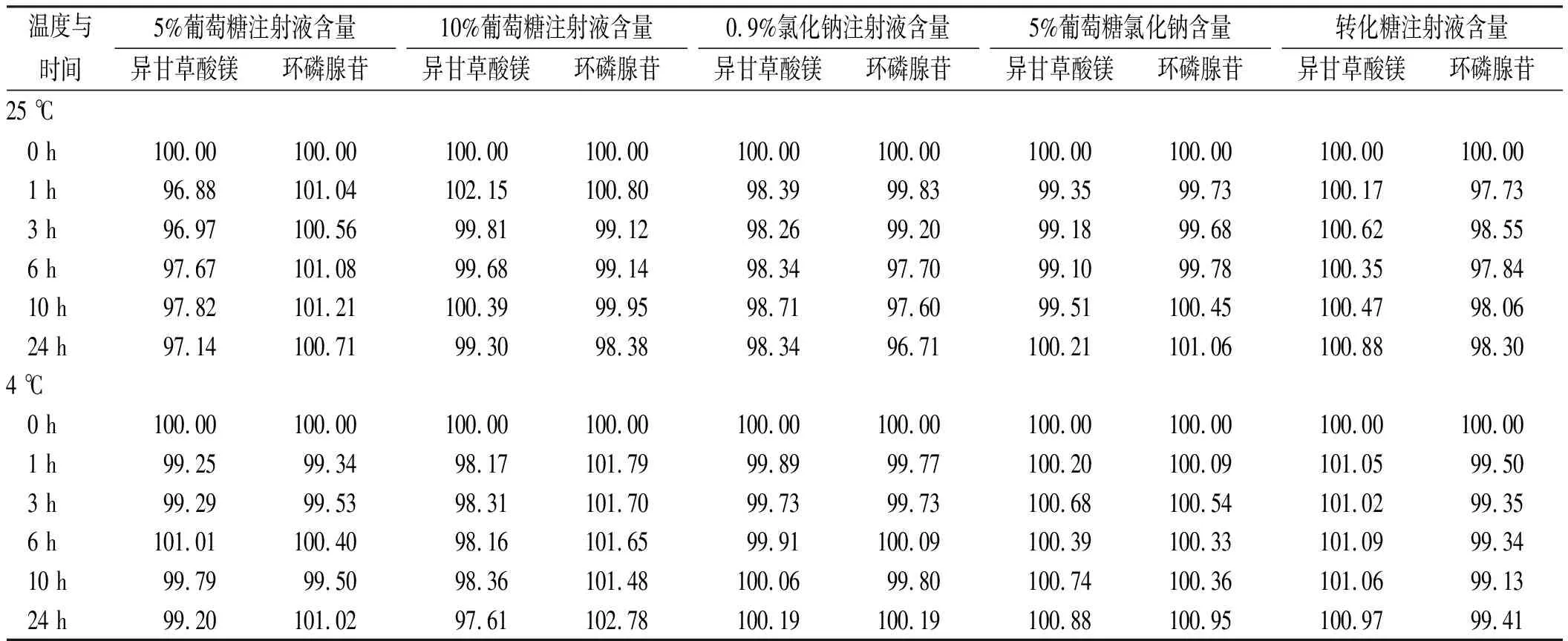
表2 不同溫度下24 h內含量變化

表3 不同溫度下24 h內不溶性微粒的變化
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
