復方巴旦仁顆粒中甘草酸的含量測定
2019-10-15 07:21:44王曉敏趙如月
中國民族民間醫藥 2019年17期
王曉敏 趙如月 周 凡
新疆華世丹藥物研究有限責任公司,新疆 烏魯木齊 830011
復方巴旦仁顆粒由巴旦仁、蜀葵子、睡蓮花、甘草浸膏等六味藥材組成,是維吾爾名醫經驗方,在臨床上主要用于寒性感冒、咳嗽痰多、氣喘、咽喉腫痛、鼻塞流涕、支氣管炎干咳、哮喘等癥的治療。維吾爾醫認為復方巴旦仁顆粒具有生濕生熱,祛除風寒,調節異常氣質,止咳化痰,溫肺止喘的功效[1]。該處方中含有的甘草浸膏,既是我國傳統中醫藥的重要組成,也是維吾爾醫藥中的常用藥味,具有補脾益氣、清熱解毒、祛痰止咳、調和諸藥等作用[2]。目前關于甘草及其提取物的化學成分和藥理作用研究相對比較充分,現代藥理學表明,甘草和甘草提取物在呼吸系統方面具有止咳、化痰、定喘以及抗呼吸道感染等作用,特別是甘草中的黃酮類成分以及甘草酸、甘草次酸在呼吸系統方面作用顯著[3-4]。復方巴旦仁顆粒為在研的中藥復方制劑,為有效控制其質量,本研究在質量標準的建立過程中選擇甘草酸作為質量控制指標之一,建立復方巴旦仁顆粒中甘草酸的含量測定方法。
1 儀器與試藥
儀器:島津LC-20AD高效液相色譜儀,FA1004分析天平(上海恒平科學儀器有限公司),CPA225D分析天平(賽多利斯科學儀器有限公司)。
試藥:甘草酸銨對照品(中國食品藥品檢定研究院,批號110731-201619,含量93.0%),復方巴旦仁顆粒(新疆華世丹藥業股份有限公司生產,批號1902160101、1902160201、1902160301;新疆華世丹藥物研究有限責任公司自制,批號20180202),乙腈、冰乙酸均為色譜純,水為超純水。
2 方法與結果
2.1 色譜條件 色譜柱為Agilent TC-C18(4.6×250 mm,5 μm);流動相:乙腈-1%冰乙酸溶液(39∶61);檢測波長:250 nm;流速:1.0 mL/min;柱溫:40 ℃;進樣量:10 μL。理論塔板數按甘草酸峰計算應不低于5000。
2.2 溶液配制
2.2.1 甘草酸銨對照品溶液 精密稱取甘草酸銨對照品26.77 mg,用50%乙腈溶解并定容至50 mL容量瓶中,搖勻,即得甘草酸銨對照品儲備液。精密量取上述溶液2 mL至10 mL容量瓶中,用50%乙腈稀釋并定容至刻度,搖勻,即得甘草酸銨對照品溶液(每1 mL含甘草酸銨約0.1 mg,折合甘草酸約為0.098 mg)。
2.2.2 復方巴旦仁顆粒供試品溶液 取本品約0.3 g,精密稱定,置具塞錐形瓶中,精密加入流動相50 mL,超聲處理20 min,取出,放冷,搖勻,用0.22 μm微孔濾膜濾過,即得。
2.2.3 陰性對照顆粒供試品溶液 按處方比例稱取除甘草浸膏以外的其他藥味,按照復方巴旦仁顆粒的生產工藝制成陰性對照顆粒,再按2.2.2項下的方法制成缺甘草浸膏的陰性對照溶液。
2.3 方法學考察
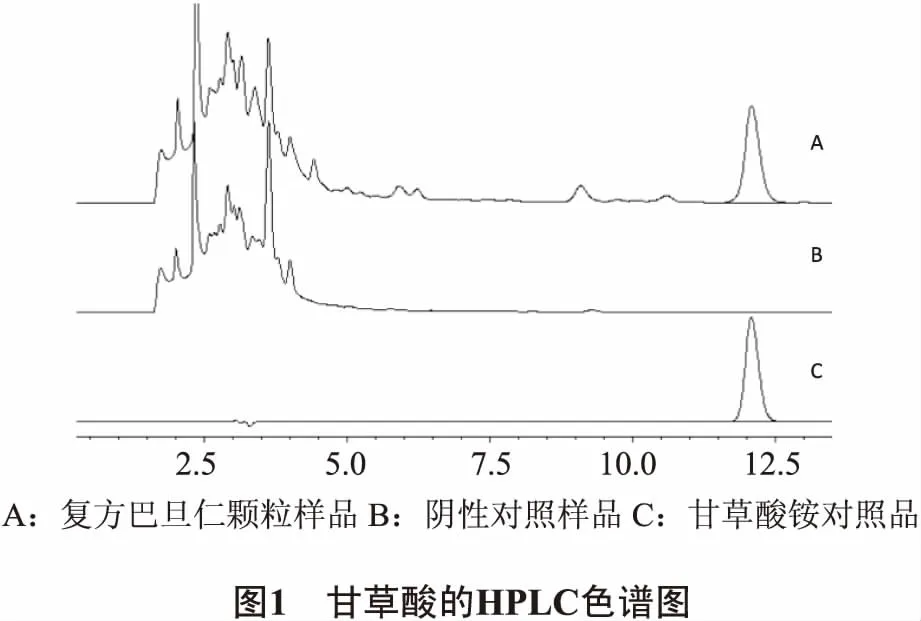
2.3.1 專屬性 精密吸取供試品溶液、陰性對照溶液、甘草酸銨對照品溶液各10μL進樣分析,記錄色譜圖(見圖1)。結果陰性對照溶液在甘草酸銨色譜峰相應位置上無干擾,可在此條件下進行樣品中甘草酸的含量測定。
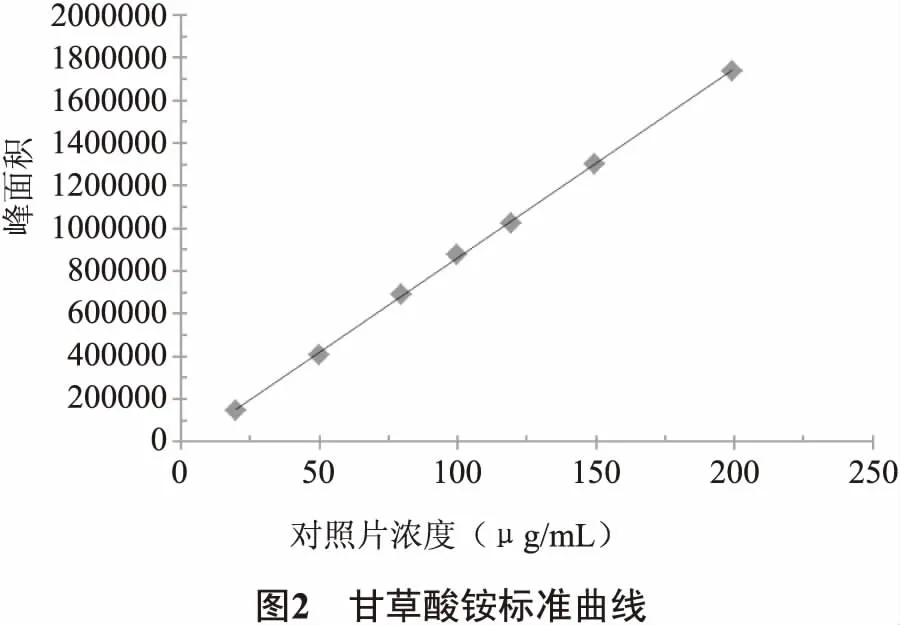
2.3.2 線性關系考察 分別精密量取甘草酸銨對照品儲備液0.4 mL、1.0 mL、1.6 mL、2.0 mL、2.4 mL、3.0 mL、4.0 mL至10 mL容量瓶中,用50%乙腈稀釋并定容至刻度,搖勻,即得線性系列溶液。取上述對照品溶液各10 μL,注入色譜儀分析測定,記錄色譜圖,以濃度為橫坐標,峰面積為縱坐標,進行線性回歸。得回歸方程Y=8873.9X-25182(R2=0.9996,n=7),見圖2。即甘草酸銨濃度范圍在19.92~199.17 μg/mL時,線性關系良好。
2.3.3 精密度 精密吸取甘草酸銨對照品溶液10 μL,重復進樣6次,結果甘草酸銨峰面積的RSD=0.05%,表明儀器精密度良好。
2.3.4 加樣回收率試驗 取已知含量的復方巴旦仁顆粒(批號20180202)適量,加入低、中、高(50%、100%、150%)系列濃度的對照品,按擬定的色譜條件測定,計算回收率,結果平均回收率102.49%(n=9,RSD=2.05%),見表1,表明方法回收率較好。
2.3.5 重復性 分別稱取同一批號復方巴旦仁顆粒(20180202)5份,按照2.2.2項下的方法制備成供試品溶液,進樣測定。結果樣品中甘草酸的平均含量為1.257%,RSD=1.49%,表明方法重復性較好。
2.3.6 溶液穩定性 取同一供試品溶液,分別于0、2、4、8、12、24 h進樣測定,結果甘草酸銨峰面積的RSD=0.08%,表明復方巴旦仁顆粒供試品溶液在24 h內穩定性良好。
2.4 樣品測定 分別稱取3批(1902160101、1902160201、1902160301)復方巴旦仁顆粒樣品,按照2.2.2項下的方法制成供試品溶液,并按2.1項下的色譜條件進行測定,計算含量。結果見表2。從該測定結果可以看出中試車間連續生產的3批樣品質量穩定,批間差異不大。
3 討論
3.1 流動相的選擇 本實驗曾用《中國藥典》2015版中甘草浸膏和甘草流浸膏的方法進行試測,結果均不理想,因此查閱相關資料進行了流動相調整[5-9]。實驗先后考察了乙腈-磷酸體系、乙腈-冰乙酸體系和甲醇-冰乙酸體系,發現乙腈-冰乙酸體系可以獲得較好的峰形且分析時柱壓更低,在保留時間相當時分離效果更好。通過調整有機相-水相比例和酸濃度,最終發現流動相為乙腈-1%冰乙酸溶液(39∶61)時,所得峰形對稱性好,樣品主峰和雜質峰可以有效分離,理論塔板數較高。

表1 樣品的回收率測定

表2 三批樣品含量測定結果
3.2 檢測波長的選擇 取甘草酸銨對照品溶液,在200~400 nm波長范圍內掃描,結果發現甘草酸銨在250.6 nm處有最大吸收,最終選擇250 nm作為液相色譜含量測定的檢測波長。
3.3 耐用性 實驗表明,當流速變化為(1.0±0.1)mL/min、檢測波長變化為(250±5)nm時,對含量測定的結果幾乎沒有影響。柱溫的改變會影響甘草酸銨主峰與周圍雜質峰的分離,經測試,柱溫40 ℃下能達到較好的分離效果。
3.4 提取條件 本實驗考察了不同超聲時間制得的樣品含量,結果顯示超聲提取20 min即可提取完全;還考察了使用不同提取溶劑配制的樣品含量,結果用流動相提取時效率高且雜質干擾少。
甘草酸為甘草浸膏中的主要成分,止咳祛痰作用明確,也是復方巴旦仁顆粒中的重要藥效成分。采用本法測定復方巴旦仁顆粒中的甘草酸含量,操作簡便,結果準確、可靠,可用于控制復方巴旦仁顆粒的質量。同時,現代分析技術與傳統民族藥的結合有利于推進民族藥的產業化和規范化,該方法的建立對完善民族藥的質量標準提供了一定參考。
