GC法檢測黃芪飲片中有機氯農藥殘留量
2019-10-23 14:28:29高艷梅趙梅
中國民族民間醫藥·下半月 2019年7期
高艷梅 趙梅
【摘 要】 目的:對黃芪飲片中有機氯農藥殘留量的可行性進行研究。方法:采用氣相色譜法,檢測器為電子捕獲檢測器(ECD),溫度 300℃;進樣口溫度 250℃,選擇程序升溫,載氣流速是1.5mL/min,恒壓模式,不分流;HP–5色譜柱(30m×0.32mm,0.25μm);樣品用丙酮、二氯甲烷提取,濃硫酸磺化。結果:標準曲線法計算農藥含量,加標回收率為94.5%~104.4%,測定結果的相對標準偏差為0.8%~1.5%,檢出限為0.061~0.175ng/mL。結論:此方法可用于測定其有機氯農藥殘留量,穩定、可靠、簡單、快速。
【關鍵詞】 氣相色譜法;黃芪飲片;有機氯農藥
【中圖分類號】R284.1 【文獻標志碼】 A【文章編號】1007-8517(2019)14-0044-03
中藥作為傳統藥物,在中國有著上千年的使用歷史。隨著中藥產品的影響力越來越大,中藥材更是受到國際高度關注。近年來受到國際貿易中綠色、技術等貿易壁壘的影響,中藥產品的出口貿易現狀不容樂觀。其中,農藥殘留是影響中藥材質量安全的主要因素,同時也是嚴重制約我國中藥產品走向國際市場的重要原因,直接影響了中藥在國際市場上的競爭力[1]。
黃芪具有補氣升陽,固表止汗,利水消腫,生津養血,行滯通痹,托毒排膿,斂瘡生肌的功效。不管是用于治病還是用于保健,都越來越受到廣大人民的喜愛,保證其質量安全,至關重要。我國中藥走向國際市場,受到農藥殘留問題嚴重阻礙作用,規范中藥材種植和管理關鍵措施,建立有效的農藥殘留量測定方法,以此來規范中藥種植源頭的農藥殘留。目前,用于測定食品和中藥材中有機氯農藥的方法主要為氣相色譜法和氣相色譜-質譜聯用法[2-3]。研究采用氣相色譜法分析檢測黃芪飲片中有機氯農藥殘留, 通過方法學考察, 本方法在實際檢測中穩定、可靠、簡單、快速。
1 儀器與材料
1.1 儀器 7890A氣相色譜儀(美國Agilent公司);KQ-300DE數控超聲波清洗器(昆山市超聲儀器有限公司);SC-3610低速離心機(安徽中科中佳科學儀器有限公司);RE-52A旋轉蒸發器(上海安亭電子儀器廠);N-EVAP-112氮吹儀(美國Organomation公司)。
1.2 材料 黃芪飲片選自醫院。滴滴涕(op-DDT,PP′-DDT,PP′-DDE,PP′-DDD)及六六六(γ-BHC,δ-BHC,α-BHC,β-BHC)(混合對照品溶液批號:SB05-068-2018),濃度:100μg/ml;五氯硝基苯(PCNB)(批號:GB05-1845-2018)等對照品都來自農業部環境保護科研監測所研制。剩下試劑均是色譜純。
2 方法與結果
2.1 色譜條件 進樣口:250℃;檢測器ECD溫度:300℃;程序升溫:初始溫度60℃,保持0.5min,以每分鐘60℃升到170℃,再每分鐘以10℃升到220℃,保持5min,再每分鐘以1℃升到240℃,再每分鐘以15℃升到280℃;載氣流速是1.5mL/min,高純氮氣(>99.999%),恒壓模式,尾吹流量是58.5mL/min,進樣量是1μL,不分流。
2.2 制備混合對照品溶液 取上述混合對照品溶液和五氯硝基苯,用石油醚稀釋制成分別為10.0μg·mL-1的溶液,作為混合對照品儲備液。取適量混合對照品儲備液,用石油醚分別稀釋成0.0、0.2、0.4、0.6、、0.8、1.0μg/mL的系列混合對照品溶液,用于線性回歸方程。
2.3 制備供試品溶液 取適量黃芪飲片,用中藥粉碎機粉碎,過三號篩,置于100mL燒杯中,稱取約2.0g,加水浸泡過夜,加6g氯化鈉,40mL丙酮,稱重后超聲處理30min,之后將30mL二氯甲烷加入其中,超聲處理15min,加入適量無水硫酸鈉,過濾,將有機相置于100mL燒杯中,靜置4h,置旋轉蒸發儀中,低溫旋轉濃縮至近干,用5mL石油醚溶解,轉移至10mL離心管中,緩緩加入1mL硫酸,渦旋離心10min[4]。取上清液,用0.22μm的微孔濾膜進行過濾,取濾液,即得;同時制備空白溶液,按2.1色譜條件進樣。
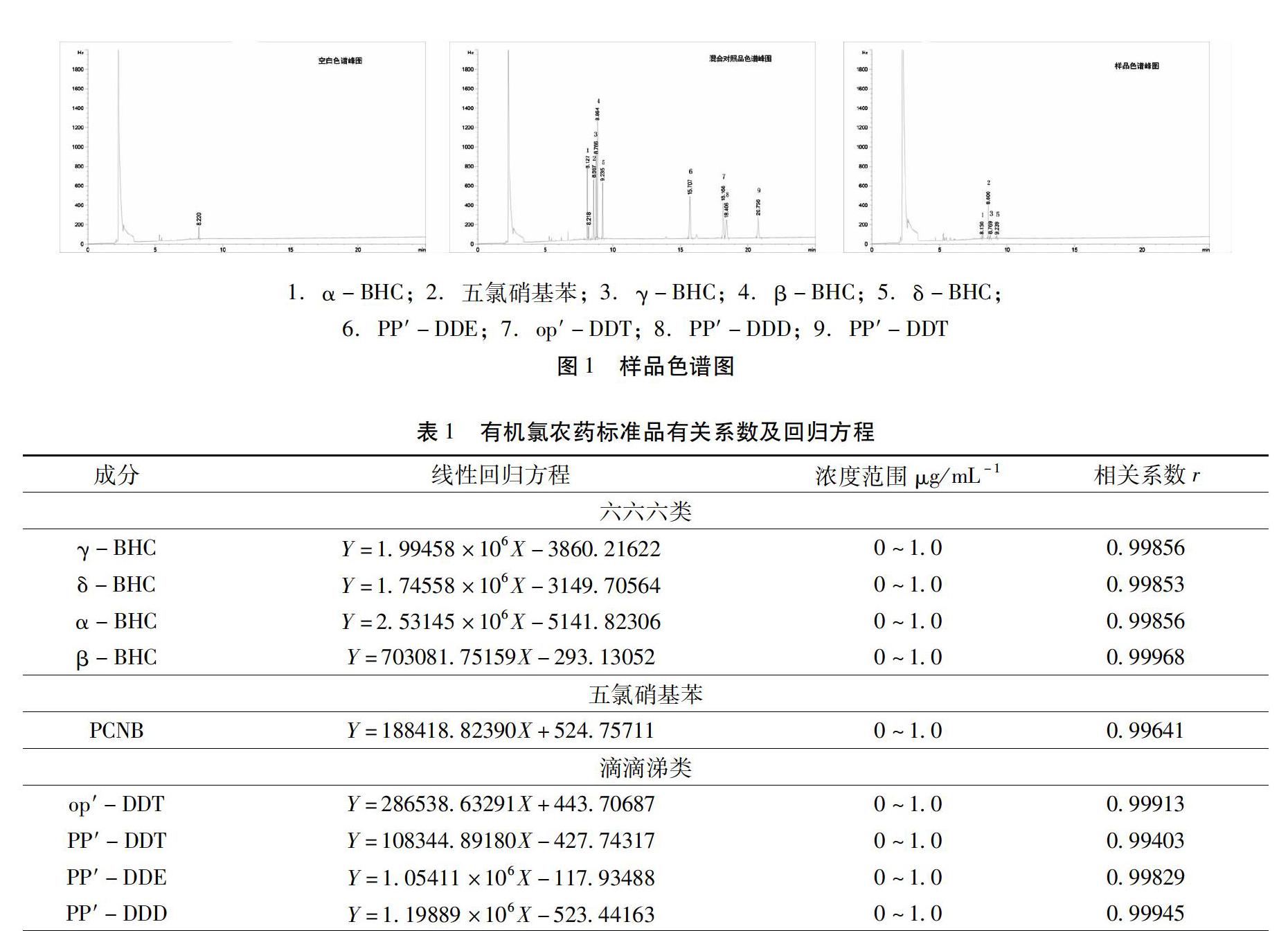
2.4 專屬性試驗 分別吸取上述2.2項下的混合對照溶液、2.3項下的供試品溶液及空白溶液,按2.1色譜條件進行檢測分析,經分析后顯示,供試品溶液色譜圖與混合對照溶液色譜圖在相對應位置存在部分相同保留時間的色譜峰,及部分六六六類和五氯硝基苯(PCNB)有殘留,滴滴涕(op-DDT,PP′-DDT,PP′-DDE,PP′-DDD)未檢出,此外可以看出,空白溶液色譜圖無干擾峰。其各成分的出峰順序如圖1所示。
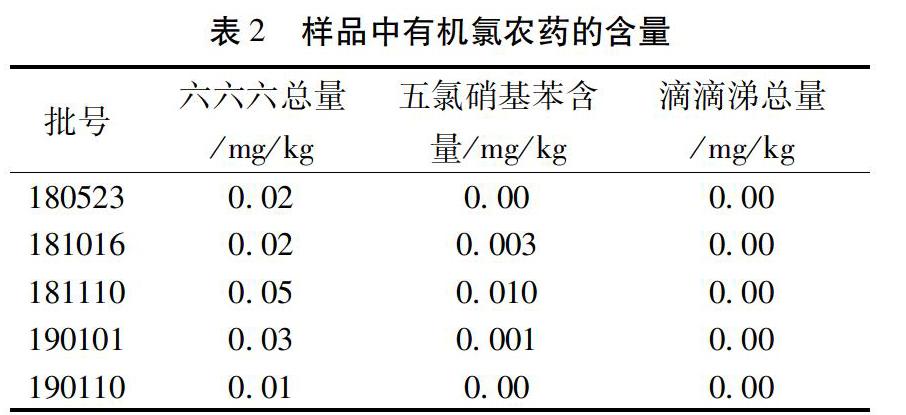
2.5 線性范圍 精密吸取1μL系列混合對照品溶液,進入氣相色譜儀,每一濃度均進行5次平行測定,以平均峰面積為縱坐標,以濃度為橫坐標,計算出線性回歸方程及相關系數。見表1,其表明混合對照品溶液線性關系良好。
2.6 精密度試驗 取同一批次供試品(批號:180523),按2.3項下的方法制備供試品溶液,按2.1色譜條件連續進樣6次,記錄峰面積。結果六六六類和五氯硝基苯峰面積的RSD為0.2%~1.0%,表明儀器精密度良好。
2.7 溶液穩定性試驗 取同一批次供試品(批號:181016),按2.3項下的方法制備供試品溶液,分別在0、1、2、4、6、8、10、12、14、16、18、20、24h時按2.1色譜條件連續進樣測定,記錄峰面積。結果六六六類和五氯硝基苯峰面積的RSD為0.8%~1.5%,表明供試品溶液在24h內較穩定。
2.8 重復性試驗 取同一批次供試品(批號:181110),按2.3項下的方法制備供試品溶液,平行制備6份,按2.1色譜條件進樣測定,結果六六六類和五氯硝基苯平均含量為0.01~0.05mg/kg,其RSD為0.5%~1.2%,表明重復性良好。
2.9 回收率試驗 精密稱取9份供試品(批號:181110),加入三種混合對照品溶液(濃度分別為0.2、0.4、0.8μg/mL),每一濃度加三份,各5mL;按2.3項下的方法制備供試品溶液,按2.1色譜條件平行進樣各5次測定。計算回收率。結果表明回收率為94.5%~104.4%,其RSD(n=6)為0.8%~1.5%。
2.10 供試品含量測定 取5個批次的黃芪飲片,按2.3項下的方法制備供試品溶液,按2.1色譜條件進樣測定,計算含量。結果見表2。
3 討論
3.1 檢出限的確定 根據測定有機氯農藥時的稱樣量、定容體積、標準物質的響應值、ECD的靈敏度、空白試樣的噪聲水平等因素來確定每個農藥成分的檢出限[5]。本方法采用的試劑為色譜純,減少雜質的干擾,提高靈敏度;采用低溫旋轉蒸發,防止有效成分被破壞或損失。配制一系列的低濃度樣品與空白樣品進行信號比較,以信噪比為3:1的量計算出能被可靠地檢測出的被測物最低濃度來確定其檢出限。
3.2 色譜分離條件的優化 有文章報道,流速為恒流模式 (1mL/min);程序升溫: 初始溫度70℃, 保持 1.0min, 以80℃/min升溫至 170℃, 以3℃/min升溫至190℃, 以0.7℃/min? 升溫至 210℃, 以 5℃/min升溫至220℃并保持10min[6]。此色譜條件用于美國安捷倫公司30m×0.32mm,0.25μm的 HP–5色譜柱上,效果很差,部分色譜峰難以分離,分離度小于1.5。本實驗對9種有機氯類農藥色譜條件進一步優化,選取三個主要因素(程序升溫、色譜柱、流速),進行色譜優化試驗,經過多種的程序升溫、流速的變化和多個型號的色譜柱(Cpsil8CB、DB-1、DB-50、DB-1701、HP-5)進行分離,綜合色譜圖分離效果,選擇最終色譜條件。本方法建立的色譜條件優點在于:第一,采用恒壓模式,峰形平穩;第二,出峰相對多的地方升溫速率慢,可以很好的分離各色譜峰,出峰相對少的地方,采用快速升溫,確保總體的保留時間較短,同時也能很好的分離色譜峰;第三,末尾采用高溫280℃,可以較好地分離出樣品中殘留的其他雜質;本方法可以用于20多種農藥殘留的分析,且分離效果很好。
3.3 結果分析 在采集5組樣品中,經測定的結果顯示,均符合《中國藥典》對黃芪藥材規定(≤0.2mg/kg),以及《藥用植物及制劑進出口綠色行業標準》對飲片、提取物及制劑、植物原料的規定(≤0.1mg/kg),有機氯農藥殘留量均很低。同時在黃芪飲片中,六六六存在3種異構體和五氯硝基苯有殘留,而相較于最低檢出限,其它含量均較低并未被檢出。然而針對于有機氯農藥殘留量來說,不同批號的黃芪藥材存在的差異較大,可能和黃芪的種植過程有關,才造成此種現象的出現,另外也可能和氣候等環境因素有關,造成農藥殘留的積集及轉化機制均需進一步研究,并積極的提供一定安全理論依據用于控制農藥殘留,保證其質量極其關鍵。
綜上,實驗建立黃芪飲片中9種有機氯類農藥殘留的毛細管氣相色譜測定法,該方法分析時間短,各農藥成分分離度良好(R>1.5),從方法的精密度和回收率來看,均能滿足實際分析的需要,并制定了快速,簡便的樣品前處理方法,這對其它中藥材有機氯類農藥殘留測定的研究具有一定的參考價值。從樣品的測定結果看,黃芪藥材中仍有少量的農藥殘留,雖然我國早已禁止生產和使用該類農藥20多年,這可能與該類農藥在環境中穩定性好、半衰期長,不易分解等有關[7]。有機氯農藥會造成慢性中毒,殘效期長,易積蓄,使用歷史長,用量大,對人體危害極大。此次研究檢測結果均符合要求,且有機氯農藥的殘留在大多數黃芪飲片中均處于國家標準范圍中。
參考文獻
[1]李慧君,張文生,吳潔珊,等.中藥材農藥殘留研究現狀[J].中國中藥雜志,2019,44(1):48-52.
[2]劉芳,歐陽慧子,柴士偉,等.GC法檢測72種中藥飲片有機氯農藥殘留量[J].中國藥房,2016,27(36):5147-5150.
[3]靳貴英,張萬青.氣相色譜-串聯質譜法測定海藻羊棲菜中的19種有機氯農殘[J].中國藥師,2017,20(12):2173-2176.
[4]靳敏,梁青青,張麗萍,等.蒙古黃芪種植土壤及藥材中重金屬和農藥殘留分析[J].中藥材,2015,38(3):454-456.
[5]萬益群,謝明勇.中草藥中有機氯農藥和擬除蟲菊酯農藥殘留量的測定[J].分析化學研究報告,2005,33(5):614-618.
[6]李烏云塔娜,云文潔.氣相色譜法測定北沙參中有機氯農藥的殘留量[J].食品安全質量檢測學報,2018,9(10):2507-2511.
[7]杜妍,宗凱.氣相色譜法檢測黃芪飲片中有機氯農藥殘留量[J].安徽醫藥學雜志,2016,20(10):1857-1860.
(收稿日期:2019-04-30 編輯:劉斌)
