可可堿、茶堿和咖啡堿的快速測定及其色譜保留行為
2019-10-29 06:38:56聶吉語湯書華姜子濤
食品科學 2019年20期
聶吉語,李 榮,*,王 穎,湯書華,譚 津,包 榮,姜子濤,2,*
(1.天津商業大學生物技術與食品科學學院,天津市食品生物技術重點實驗室,天津 300134;2.天津天獅學院食品工程學院,天津 301700)
生物堿是一類于植物體內廣泛存在的含氮雜環的堿性天然有機物,其中絕大部分具有重要的醫療價值[1-5]。可可堿、茶堿和咖啡堿是天然甲基黃嘌呤類生物堿,具有能夠使中樞神經系統、心臟和骨骼肌興奮,血管達到舒張狀態、平滑肌得到松弛和利尿等生理作用[6]。三者母體結構相同,但甲基取代基的位置和數目存在差異。可可堿與茶堿分別為3,7位和1,3位的二甲基取代,二者為同分異構體,而咖啡堿為1,3,7位的三甲基取代。作為一類具有顯著生物活性作用的生物堿,可可堿、茶堿和咖啡堿普遍存在于茶葉、咖啡、可可和糖果等食品中[7-8]。近年來,其制品在食品工業中更是得到了廣泛應用,并且國標中明確規定咖啡堿可用于可樂型飲料。但如果對以上3 種物質的攝入量未加控制而過量食用,則會產生一定的毒副作用[9-11],因此建立一種簡便、快速檢測可可堿、茶堿和咖啡堿的方法非常必要。
高效液相色譜(high performance liquid chromatography,HPLC)法是測定可可堿、茶堿和咖啡堿的最常用的方法[12-14],檢測一般采用C18反相色譜柱[15-18]。王增盛[19]采用μ Bondapak C18柱,以N-二甲基甲酰胺、甲醇、冰醋酸溶液為流動相,同時測定了茶葉中的咖啡堿、茶堿和可可堿的含量;梁燕妮[20]采用C18柱,以甲醇和水為流動相,梯度洗脫測定了不同廠家六堡茶中可可堿、茶堿、咖啡堿的含量;劉志彬等[21]采用Symmetry C18柱,以乙腈和乙酸溶液為流動相,梯度洗脫測定了10 種典型武夷巖茶葉片及沖泡茶湯中可可堿、茶堿和咖啡堿的含量;陳靜等[22]采用Unitary C18柱,以甲醇和0.5%甲酸溶液為流動相,同時測定了可可堿和咖啡堿的含量。
以上報道中雖可同時檢測1~3 種生物堿,但流動相條件復雜,含有酸或鹽且需要調節pH值,以及需要梯度洗脫等。這些分離分析方法均建立在以硅膠為基質的色譜柱基礎上,其游離的硅羥基會導致非特異性吸附,可可堿、茶堿和咖啡堿等一些極性較強的生物堿色譜峰會嚴重拖尾,甚至會因強吸附而不能洗脫[23],從而使分離效率降低,同時使色譜柱的使用壽命大幅縮短。而作為近年發展起來的新型色譜填料,鈦膠基質固定相既可以作為Lewis酸,也可以作為Lewis堿,較傳統的硅膠基質固定相優勢明顯。并且其不存在硅膠固定相的“第2效應”,同時在pH 1~14范圍內均非常穩定,對生物樣品中帶有磷酸基團的物質還具有特殊的吸附能力,分離選擇性也很高,而硅膠固定相則僅適用于pH 3~9的范圍內。
本實驗采用鈦膠基質色譜固定相建立的反相HPLC可同時分離,分析結構相似的3 種生物堿——可可堿、茶堿和咖啡堿,該法流動相成分簡單且不含鹽,僅為5%甲醇溶液,且采用等度洗脫,檢測效率高、分離靈敏度高、檢出限低、穩定性好。將此法應用于6 種飲料樣品中可可堿、茶堿和咖啡堿的測定,在重復性、回收率等方面均表現優異,結果令人滿意。另外,本實驗也研究了3 種生物堿保留的熵、焓和吉布斯自由能,從熱力學的角度探討3 種生物堿在HPLC上的保留行為。
1 材料與方法
1.1 材料與試劑
可可堿標準品(99%)、茶堿標準品(99%)、咖啡堿標準品(99%) 上海源葉生物科技有限公司;甲醇(色譜級) 美國Sigma公司;6 種飲料樣品 市購。
1.2 儀器與設備
1200系列HPLC儀 美國安捷倫公司;Sachtopore-RP色譜柱(250 mm×4.6 mm,5 μm,300 ?) 美國Zirchrom公司;Lambda 25紫外-可見分光光度計 珀金埃爾默儀器有限公司;H2050R-1離心機 長沙湘儀離心機儀器有限公司;AUY120萬分之一天平 日本島津公司。
1.3 方法
1.3.1 標準溶液的配制
1.0 mg/mL可可堿標準儲備液:準確稱取0.100 0 g可可堿標準品,用超純水溶解并定容至100 mL容量瓶中;2.0 mg/mL茶堿標準儲備液:準確稱取0.100 0 g茶堿標準品,用超純水溶解并定容至50 mL容量瓶中;3.0 mg/mL咖啡堿標準儲備液:準確稱取0.150 0 g咖啡堿標準品,用超純水溶解并定容至50 mL容量瓶中,以上各儲備液均置于4 ℃冰箱保存備用。
1.3.2 樣品處理
飲料樣品:準確量取5 mL已提前超聲脫氣20 min的樣品,稀釋2 倍,過0.45 μm微孔濾膜,進行HPLC分析。固體飲料樣品:準確稱取0.500 0 g樣品,加水溶解并轉移至25 mL容量瓶中定容,在60 ℃條件下采用超聲波提取30 min,隨后在4 000 r/min條件下離心10 min,上清液過0.45 μm微孔濾膜,進行HPLC分析。
1.3.3 色譜條件
流動相為水-甲醇(95∶5,V/V),等梯度洗脫;流速1.0 mL/min;進樣量20 μL;檢測波長273 nm;柱溫60 ℃。
1.3.4 標準曲線的繪制
將可可堿的標準儲備液稀釋至500、100、50、20、10 μg/mL和5
μg/mL,茶堿的標準儲備液稀釋至1 000、500、100、50、20、10 μg/mL和5 μg/mL,咖啡堿的標準儲備液稀釋至1 200、600、120、60、24、12 μg/mL和6 μg/mL,取20 μL進行HPLC分析,以各標準品的質量濃度(μg/mL)為橫坐標,色譜峰的峰面積為縱坐標繪制標準曲線,計算回歸方程。不同質量濃度的混合標準溶液由上述各標準溶液以相似水平配制而成,質量濃度由低到高依次編號為1~7。
1.3.5 熱力學參數的測定
參考文獻[23-25],并按照1.3.3節色譜條件分別在30、45、50、55 ℃和60 ℃測定可可堿、茶堿和咖啡堿的熱力學參數。
2 結果與分析
2.1 流動相中甲醇比例的確定
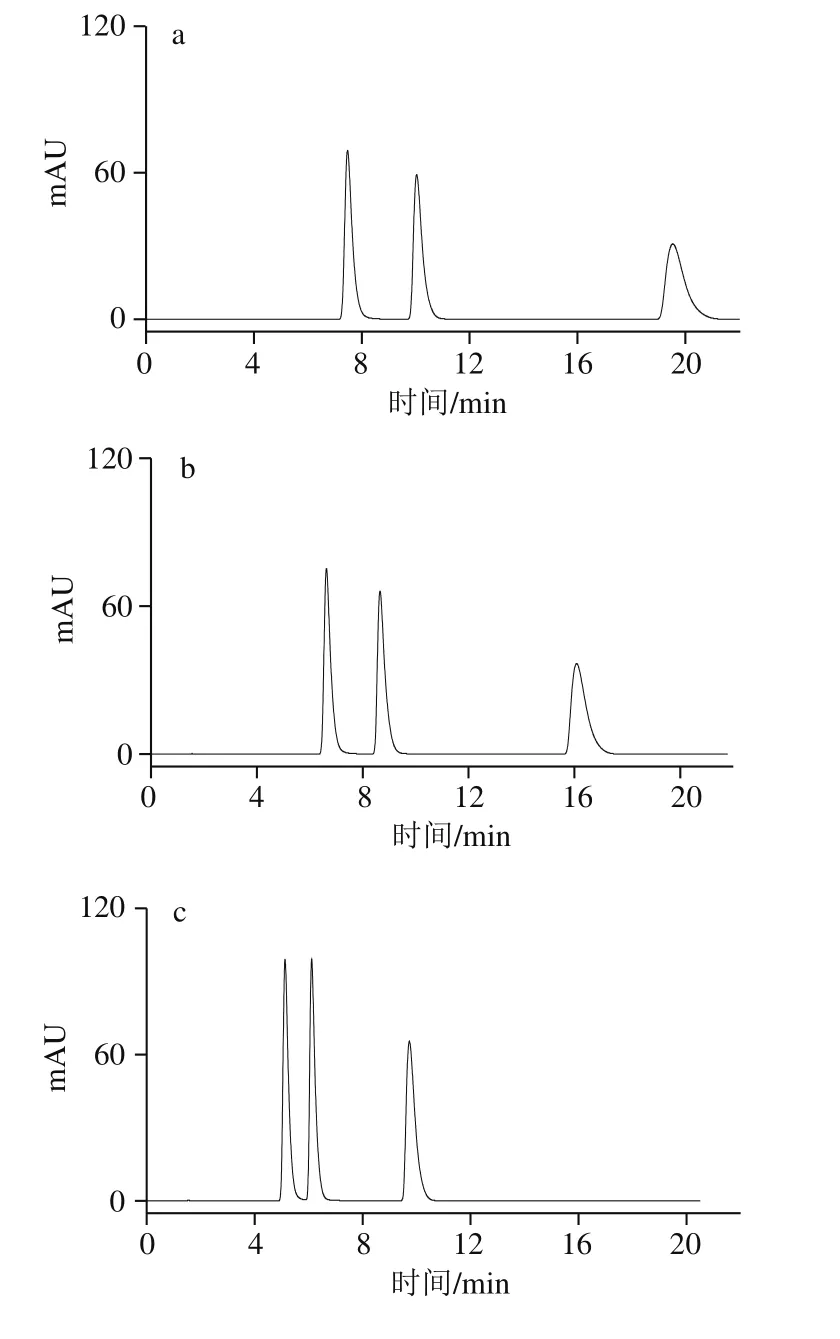
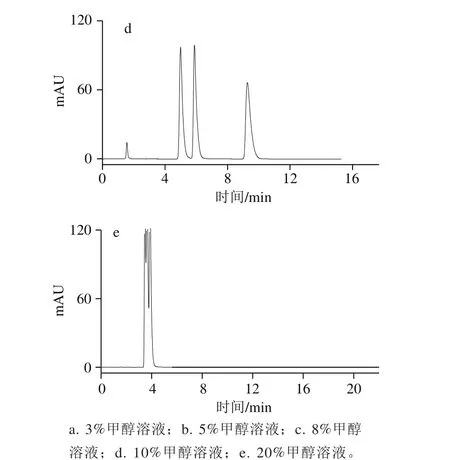
圖1 不同體積分數甲醇溶液的HPLC圖Fig. 1 HPLC profiles with different proportions of methanol in mobile phase
本實驗中流動相僅為甲醇和水,配制甲醇溶液體積分數分別為3%、5%、8%、10%和20%,按照1.3.3節色譜條件進樣分析,如圖1所示。隨著流動相中甲醇溶液體積分數的增大,流動相的極性變小,可可堿、茶堿和咖啡堿的保留時間也逐漸減小。保留時間短,雖然能夠使分析速度加快,但3 種生物堿的分離效果也會發生比較大的變化。當甲醇溶液體積分數為8%和10%時,可可堿和茶堿的分離度減少,當甲醇溶液體積分數達到20%時,可可堿、茶堿和咖啡堿三者已經不能實現完全分離。且它們的保留時間僅有4 min左右,較短的保留時間容易造成樣品制備過程中的雜質、保留較弱的非極性或弱極性物質對實驗結果產生干擾。但當甲醇溶液體積分數過低,僅為3%時,咖啡堿的保留時間與前兩者相比又過長,浪費分離時間,且存在溶質峰展寬的現象。因此,最終選擇流動相中甲醇溶液體積分數為5%,可達到各物質在基線分離的情況下,保留時間相對較短。
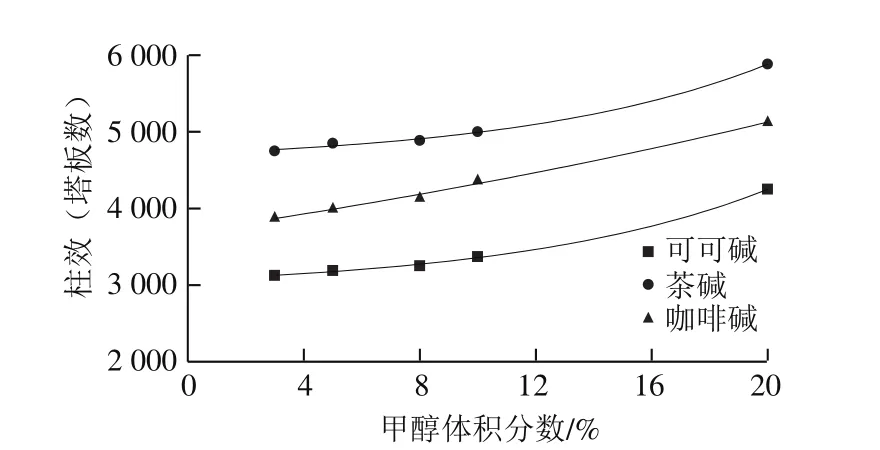
圖2 甲醇體積分數對柱效的影響Fig. 2 Effect of methanol proportion in mobile phase on number of plates
隨著流動相中甲醇溶液體積分數的增大,半峰寬也隨之減小(圖1),在同一保留時間下,半峰寬越小,柱效越高[26-27]。如圖2所示,隨著甲醇體積分數的增大,3 種堿的柱效也隨之增大;在同一甲醇體積分數下,柱效由大到小為茶堿>咖啡堿>可可堿。
2.2 柱溫的影響
2.2.1 柱溫的選擇
圖3 不同柱溫HPLC圖Fig. 3 HPLC profiles at different column temperatures
按照1.3.3節色譜條件,分別調節柱溫為30、45、50、55 ℃和60 ℃進樣分析,如圖3所示。柱溫對可可堿、茶堿和咖啡堿3 種物質的保留時間和分離度也有較大的影響,但柱溫對三者的影響稍低于流動相中甲醇溶液體積分數的影響。隨著柱溫的升高,3 種物質的保留時間也越來越小,可可堿和茶堿的保留時間減小幅度不大,咖啡堿的保留時間減少明顯,說明它們在鈦膠反相色譜柱上的保留過程是放熱的,且熱效應也有所不同。同時,增加柱溫可以減小柱壓,峰形變好,在60 ℃以上,鈦膠柱更能發揮其優良的色譜性能,然而甲醇的沸點(64.5 ℃)限制本研究最終選擇柱溫為60 ℃而不宜更高。
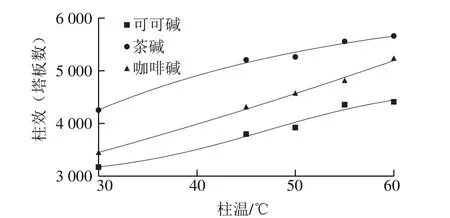
圖4 柱溫對柱效的影響Fig. 4 Effect of column temperature on number of plates
隨著柱溫的升高,可可堿、茶堿和咖啡堿的半峰寬呈減小趨勢,表明柱效不斷提高。如圖4所示,隨著柱溫的升高,3 種堿的柱效也隨之增大;在同一柱溫下,柱效由大到小為茶堿>咖啡堿>可可堿。
2.2.2 熱力學參數測定結果
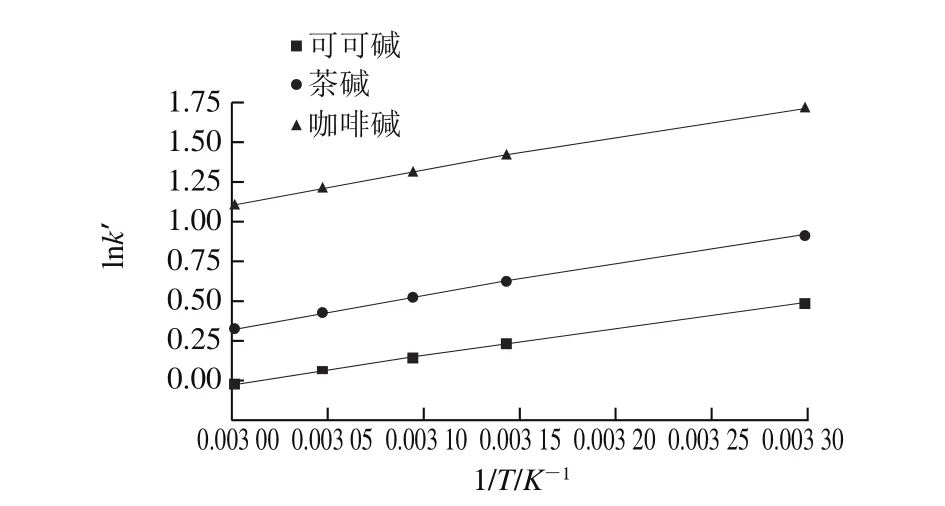
圖5 不同柱溫可可堿、茶堿和咖啡堿的Van’t Hoff曲線Fig. 5 Van’t Hoff plots for theobromine, theophylline and caffeine at different column temperatures

表1 可可堿、茶堿和咖啡堿的回歸參數Table 1 Regression parameters for theobromine, theophylline and caffeine
按照1.3.5節方法在30~60 ℃條件下對可可堿、茶堿和咖啡堿進行分析,如圖5和表1、2所示。由圖5和表1可知,在不同溫度下,可可堿、茶堿和咖啡堿在鈦膠色譜柱上的分離效果呈現良好線性,表明在本實驗所選定的溫度范圍內其在鈦膠柱上具有一致的分離機制。

表2 可可堿、茶堿和咖啡堿在333.15 K的熵、焓和吉布斯自由能Table 2 Standard entropy (ΔS0), enthalpy (ΔH0) and Gibbs free energy(ΔG0) of theobromine, theophylline and caffeine at 333.15 K
鈦膠色譜柱對可可堿、茶堿和咖啡堿的分離是由ΔG0決定的,而ΔG0又與ΔH0和ΔS0有關。由表2可知,3 種生物堿的ΔH0和ΔS0均為負值,又根據文獻[25]可知:當ΔG0為正值時,表明主要由ΔS0來驅動分析物的保留行為;當ΔG0為負值時,說明此時ΔH0在它們的保留行為中起主要作用。
熵代表了體系的混亂程度,當分析物被固定相吸附時體系混亂度較小,因為此時被吸附的物質傾向于較整齊的排列;而當分析物從固定相上解吸下來時,此時被吸附的物質趨向于做雜亂無章的布朗運動,因此體系混亂度增大[28]。由表2可以看出,3 種生物堿吸附與解吸的總熵變ΔS0為負值,并且ΔS0的順序為茶堿<可可堿<咖啡堿,說明在解吸過程中茶堿的混亂度減少最多導致其最不易被解吸,從熵變角度表明茶堿的吸附能力最強,咖啡堿的吸附能力最弱,保留順序為咖啡堿、可可堿、茶堿。
焓代表了體系的熱效應,當體系為吸熱過程,焓值升高;當體系為放熱過程,焓值降低。由表2可以看出,3 種生物堿的總焓變ΔH0為負值,說明溶質在HPLC中的保留過程為放熱過程[29-30],即吸附的貢獻大于解吸的貢獻,并且ΔH0的順序為咖啡堿<茶堿<可可堿,從焓變角度表明咖啡堿的吸附能力最強,可可堿的吸附能力最弱,保留順序為可可堿、茶堿和咖啡堿。
當分析物從流動相進入固定相為熱力學自發過程時ΔG0為負值,ΔG0越小,表明分析物越容易從流動相轉移到固定相中,即分析物在固定相上的保留會相對更強。可可堿、茶堿和咖啡堿的ΔG0均為負值,且依次減小,即咖啡堿在固定相中保留最強,茶堿次之,可可堿保留最弱,這也說明3 種堿在鈦膠固定相中的保留行為是由焓變ΔH0驅動的,實驗結果也表明焓變的數值遠大于熵變的數值,從熱力學角度闡明了分析物的保留機理。
2.3 流速的選擇
考察流速分別為0.5、0.8、1.0、1.2 mL/min和1.4 mL/min時的影響,按照方法1.3.3節色譜條件進樣分析,如圖6所示。流動相的流速對化合物的保留時間有較大影響。隨著流速的增大,可可堿、茶堿和咖啡堿的色譜峰前移,保留時間均縮短,同時其半峰寬也變小。而對于分離度而言,在5 種流速下,3 種生物堿均達到了完全分離,即分離度大于1.5。當流速小于1.0 mL/min時,保留時間過長;但當流速達到1.2 mL/min特別是1.4 mL/min時,可可堿的保留時間偏小,同時在增大流速時會導致柱壓明顯增加,從而大大降低色譜柱的使用壽命,不利于色譜儀的正常使用。因此,流動相的流速最終確定為1.0 mL/min。
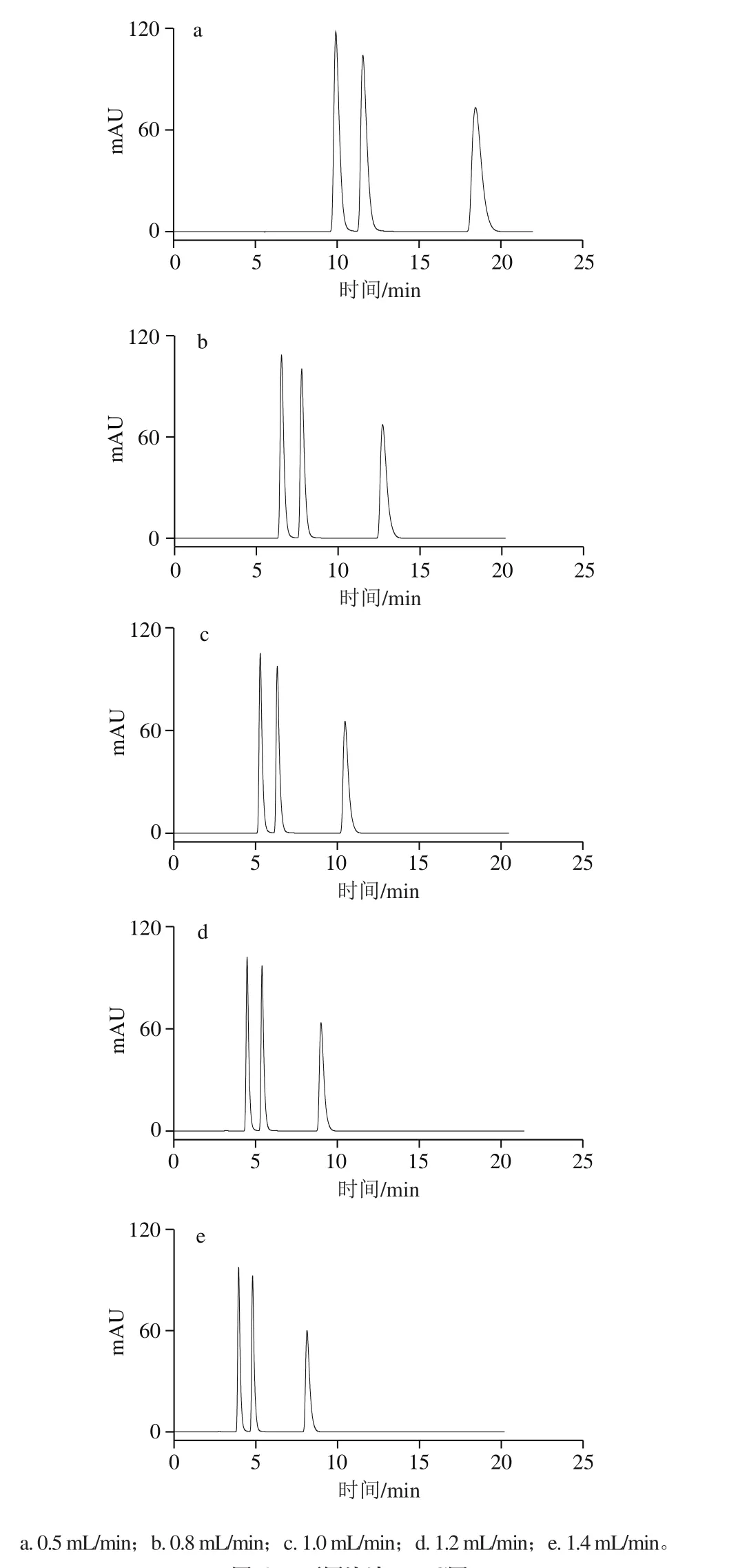
圖6 不同流速HPLC圖Fig. 6 HPLC pro fi les at different flow rates
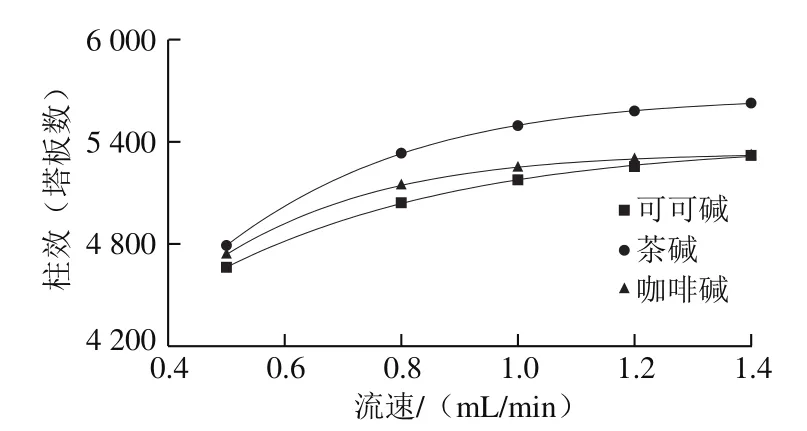
圖7 流速對柱效的影響Fig. 7 Effect of flow rate on number of plates
同樣,在流速不斷增大的情況下,理論塔板數也不斷升高,即柱效呈現增大的趨勢。如圖7所示,在同一流速下,柱效由大到小為茶堿>咖啡堿>可可堿(1.4 mL/min除外);在流速為1.4 mL/min時,可可堿和咖啡堿的柱效幾乎一致。
綜合考慮以上因素,最終確定HPLC分離分析可可堿、茶堿和咖啡堿的最佳條件為流動相水-甲醇(95∶5,V/V),柱溫60 ℃,流速1.0 mL/min,檢測波長273 nm,在此條件下三者混標與單標的HPLC圖見圖8。
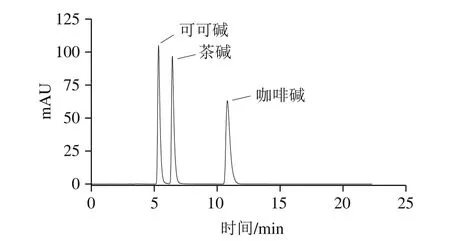
圖8 可可堿、茶堿和咖啡堿的HPLC圖Fig. 8 HPLC profiles of theobromine, theophylline and caffeine
2.4 檢出限及線性范圍結果
按照1.3.4節方法進行實驗,測定可可堿、茶堿和咖啡堿的線性范圍。結果顯示,可可堿、茶堿和咖啡堿三者分別在質量濃度范圍為5~500、5~1 000 μg/mL和6~1 200 μg/mL時,標準曲線線性良好,其回歸方程、相關系數和檢出限如表3所示。
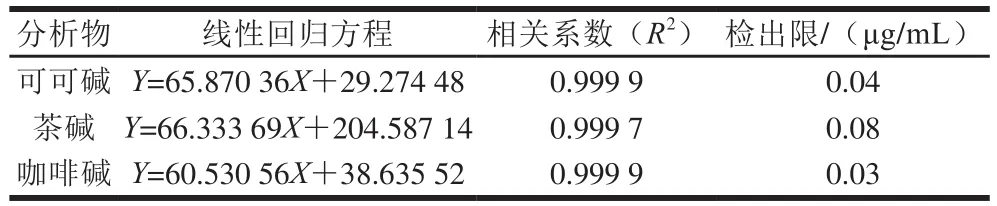
表3 可可堿、茶堿和咖啡堿的線性回歸方程、相關系數和檢出限Table 3 Linear regression equations, correlation coefficients and LODs of theobromine, theophylline and caffeine
2.5 方法精密度和準確度結果
從1.3.4節中選取3號和4號混合標準溶液在最優色譜條件下重復進樣9 次,計算得相對標準偏差即精密度小于0.23%。將可可堿、茶堿和咖啡堿加入需要測定的6 個飲料樣品中進行加標回收實驗,如表4所示,6 種樣品的色譜圖見圖9。由表4可知,可可堿、茶堿和咖啡堿的回收率范圍分別為99.0%~102.0%、96.7%~106.7%和99.4%~102.3%。由圖9可知,本法可實現對可可堿、茶堿和咖啡堿的同時分離檢測。
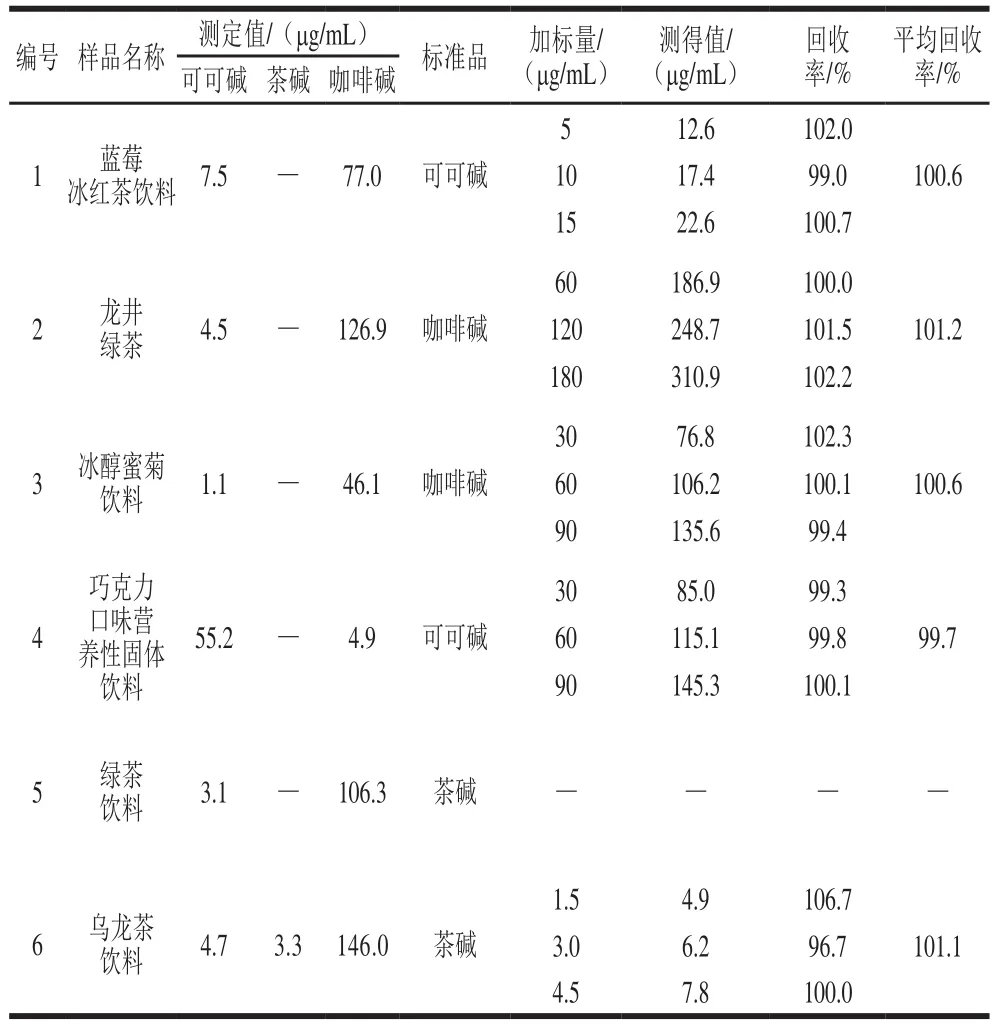
表4 實驗結果和回收率Table 4 Results and recoveries
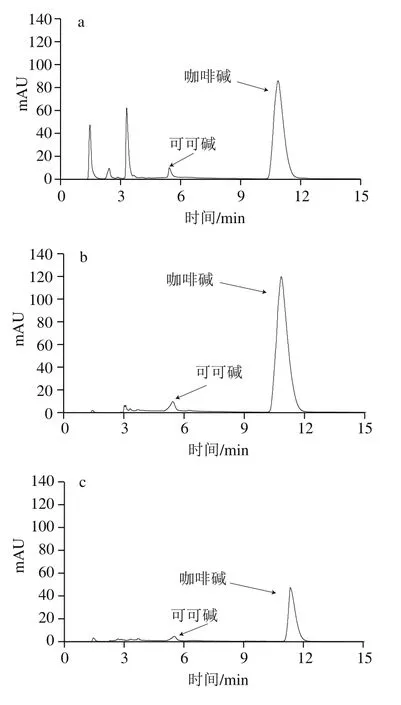
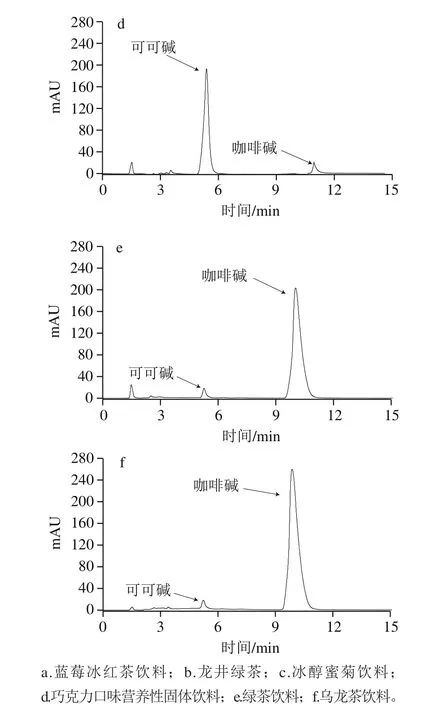
圖9 樣品HPLC圖Fig. 9 HPLC profiles of samples
3 結 論
鈦膠基質色譜固定相在天然甲基嘌呤類生物堿如可可堿、茶堿和咖啡堿的快速、同時分析檢測中展現出了優異的色譜性能,檢測結果顯示色譜峰峰形較好,可達到理想的測定效果,且其在線性范圍、檢出限以及加標回收率等方面均可達到分析要求。此外,HPLC條件(尤其是流動相)在較大程度上影響化合物的分離,流動相的試劑種類、梯度比例以及酸溶液的種類和濃度均可影響分離效果。在本實驗提出的色譜條件中,流動相中甲醇的比例僅占5%,無需添加任何緩沖鹽,采用等度洗脫簡便、快速。同時,可以避免堿性物質的不可逆吸附以及色譜柱的壽命隨使用而快速縮短等弊端,充分體現了綠色高效、保護環境、節約資源等優點,適于推廣普及。
