1例兒童自身免疫內分泌腺病綜合征Ⅲ型診治分析的報道
2019-10-30 02:44:35符蕓瑜楊銳
中國當代醫藥 2019年24期
符蕓瑜 楊銳
[摘要]兒童自身免疫內分泌腺病綜合征在臨床上較為少見,一旦出現,容易誤診、漏診,如不及時明確診斷、及時治療,往往會對整個疾病的診療過程造成一定延誤。本文對2016年9月入院的1例兒童自身免疫內分泌腺病綜合征Ⅲ型患者進行分析與總結,探討自身免疫內分泌腺病綜合征的診斷、治療,結合實際的發病及診療過程,討論如何對該病作出診斷、分型及相應的治療;提出該病需要長達數十年的隨訪,可能出現分型的改變,以便及時給予相應的診斷及治療措施。
[關鍵詞]自身免疫腺病;1型糖尿病;兒童;Graves病;綜合征
[中圖分類號] R593.2? ? ? ? ? [文獻標識碼] A? ? ? ? ? [文章編號] 1674-4721(2019)8(c)-0192-03
[Abstract] Children autoimmune polyendocrinopthy syndrome was rarely seen in the clinic, once it appeared, it might be easily missed and misdiagnosed, if not diagnosed and treated in time, the whole process of diagnosis and treatment will be delayed. One case of children autoimmune polyendocrinopthy syndrome type Ⅲ admitted in September 2016 was analyzed and summarized in this article, the objective was to probe into the diagnosis and treatment of the autoimmune endocrine adenopathy syndrome, combining with the actual paroxysm and treatment course, the diagnosis, classification and relevant treatment were discussed. This article also proposed that the disease needed follow-up for more than ten years, and there might exist the possibility of classification changes, in order that corresponding diagnosis and treatment measures could be given in time.
[Key words] Autoimmune adenosis; Type 1 diabetes mellitus; Children; Graves disease; Syndrome
自身免疫內分泌腺病綜合征(autoimmune polyendocrinopthy syndrome,APS)是臨床上的罕見病,兒童發病、診斷更是少之甚少。臨床上大多患者首診見于其他專科,隨著該病患病率的增高及對疾病認識水平的提高,最終常由內分泌專科確診、治療。APS是指由于自身免疫因素引起或導致2個或2個以上內分泌腺體功能異常的綜合征,除此之外,該病還可以累及其他非內分泌系統,如消化系統、血液系統、神經系統等,大多數病變表現為腺體功能低下,也可見功能亢進;各腺體的病變可以同時發生,也可以相繼發生,甚至相差數十年發生。現報道1例兒童自身免疫內分泌腺病綜合征Ⅲ型病例的診治分析。
1 病例資料
患兒男,11歲,因“口干、多飲、多尿、消瘦1個月”于2016年9月來院就診,1個月來體重減輕6.5 kg,查空腹指尖血糖>33.3 mmol/L,診斷為“糖尿病”收入院治療。既往病史:患者于2016年5月起出現消瘦,頸前腫物,于南方醫科大學珠江醫院就診,檢測甲狀腺功能提示:游離三碘甲狀腺氨基酸:36.06 pmol/L,游離甲狀腺素:64.63 pmol/L,促甲狀腺激素:0.01 mIU/L,促甲狀腺素受體抗體:13.2 IU/L,抗甲狀腺球蛋白抗體:13.91 kU/L,甲狀腺過氧化物酶抗體:117.5 kU/L。診斷“甲狀腺功能亢進癥”,予甲巰咪唑100 mg/d。此后每月定期復查甲功、血常規、肝功能,抗甲亢治療期間血常規及肝功能未見明顯異常,甲狀腺功能指標逐漸改善。既往無其他特殊病史。否認類似家族遺傳病史。
入院查體:體溫(T)36.2℃,脈搏(P)116次/min,呼吸(R)20次/min,血壓(BP)123/66 mmHg。發育正常,營養中等,形體消瘦,皮膚、口唇干燥,無黑棘皮、痤瘡、紫紋,全身淺表淋巴結未觸及腫大。雙眼眼球突出,眼裂增寬,Joffroy征(-)、vonGreafe征(-)、Stellwag征(-)、Mobius征(-),球結膜無充血和水腫,鞏膜無黃染。雙側甲狀腺Ⅲ度腫大,無壓痛,未聞及收縮期動脈雜音。呼吸略促,雙肺部呼吸音粗,未聞及干濕性啰音。叩診心界無擴大,心率(HR) 116次/min,律齊,各瓣膜聽診區未聞及病理性雜音。腹軟,無壓痛及反跳痛,肝、脾肋下未觸及,腎區無叩擊痛。雙下肢無水腫。雙股動脈、腘動脈、背動脈波動正常,四肢淺感覺正常。余無特殊。
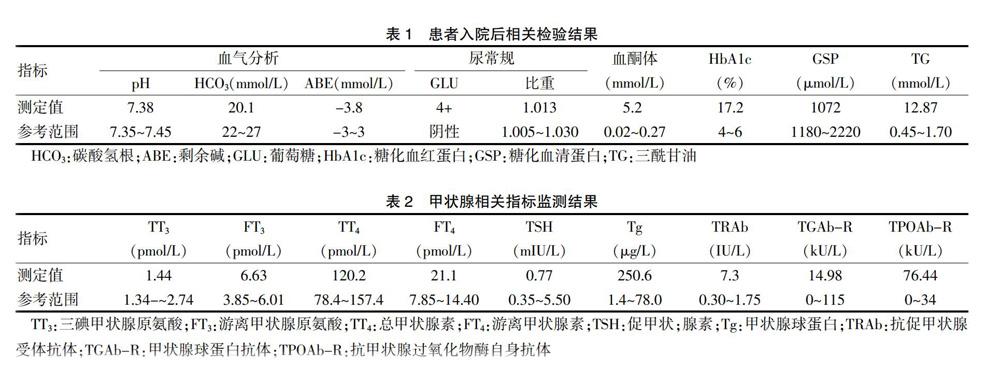
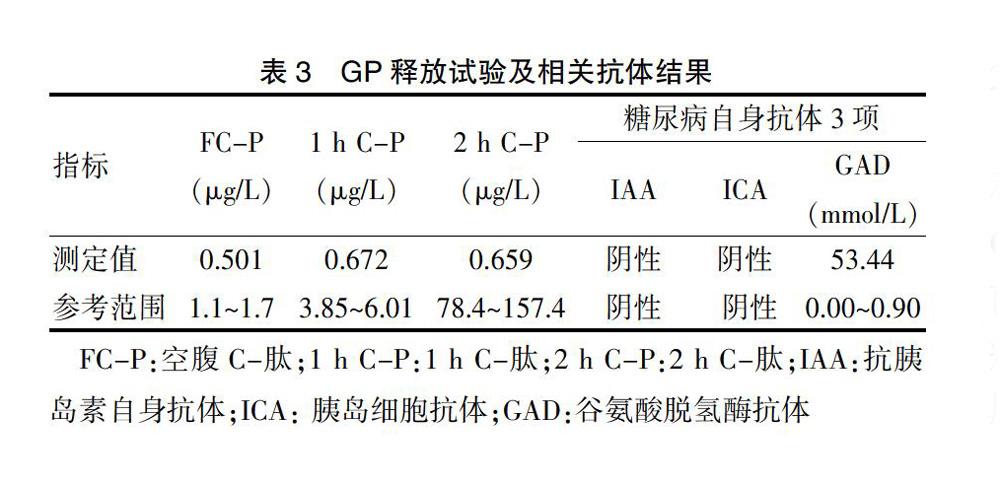
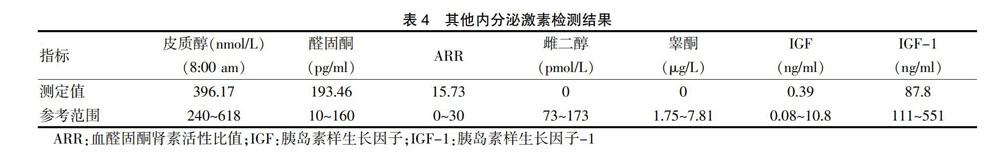
實驗室檢查結果見表1,表中未列出的血常規、肝功能、腎功、電解質均正常。同時行多內分泌腺體功能檢查,結果見表2~4,其中未列出的24 h尿游離皮質醇、甲狀旁腺激素、孕酮、泌乳素、促卵泡雌激素、促黃體生成素均正常。免疫系統檢查:風濕指標:抗鏈球菌溶血素“O”(ASO):270 kU/L(0~100 kU/L),類風濕因子(RF)正常。抗核抗體譜、血管炎指標八項、抗中性粒細胞胞漿抗體、抗磷脂綜合征指標均正常,該患者所有檢驗、檢查結果均出自南方醫科大學珠江醫院。
2 治療及轉歸
患兒為男性兒童,此次因糖尿病入院,起病急、病程短,自發酮體傾向,C-P水平差,糖尿病抗體3項中GAD陽性,明確1型糖尿病。患者在發生1型糖尿病前出現甲狀腺功能亢進癥,進一步檢查明確Graves病;根據完善的相關實驗室檢查結果可暫時除外腎上腺皮質病變以及其他免疫系統疾病,綜合考慮該患者為自身免疫多發內分泌腺病綜合征APSⅢ型:T1DM,Graves病;1型糖尿病 糖尿病酮癥。給予小劑量胰島素靜脈點滴、補液消酮、糾正電解質紊亂、改善循環和對癥治療,入院第2天復查酮體消除后給予胰島素泵皮下泵入控制血糖、補液、糾正電解質紊亂、抗甲亢、改善循環和對癥治療,血糖逐漸平穩,口干、多飲、多尿、乏力癥狀消失。后改為門冬胰島素30筆芯皮下注射聯合午餐時口服二甲雙胍片0.5 g控制血糖,根據血糖波動情況調整胰島素劑量。全天血糖波動:4.6~18 mmol/L。入院后繼續口服甲巰咪唑10 mg,2次/d。
經治療12 d后病情穩定出院,出院時全天血糖波動:3.5~11.3 mmol/L。1個月后隨訪,患者無口干、多飲、多尿、乏力癥狀,體重增加3.5 kg,血糖較前下降、平穩,波動于4.5~10.6 mmol/L。囑其繼續長期應用胰島素,暫時聯合二甲雙胍片控制血糖,繼續口服甲巰咪唑抗甲亢治療,定期復查血糖、甲狀腺功能,定期隨診。
3 討論
APS是指2個或2個以上的內分泌腺體因自身免疫功能缺陷而發生功能受損為主要表現的一系列綜合征,可以累及非內分泌腺器官[1]。APS患者表現為多器官功能障礙相互影響,其中多數表現為器官(或細胞)功能減退或衰竭,個別器官(或細胞)表現為功能亢進。根據各個器官受累情況,APS既往分型為4型,具體分型如下。
3.1 APSⅠ型
這一型以往也稱為黏膜皮膚念珠菌內分泌病。主要有以下4種和(或)病變,腎上腺皮質功能減退(AI)或腎上腺皮質自身抗體陽性﹑甲狀旁腺功能減低(HP)﹑慢性皮膚黏膜念珠菌感染三者中至少兩種病變。有些患者僅有其中一種內分泌病變,或者四種均有,但這四種病變起病時間各可相距十到數十年。除此之外,該型的患者還可以有垂體炎﹑多囊卵巢﹑禿發﹑白癜風﹑活動性肝炎,罕見的有類風濕關節炎﹑干燥綜合征[2-4,7]。
3.2 APSⅡ型
本型是AI并自身免疫性甲狀腺疾病(AITD)和(或)1型糖尿病[5]。該型不伴有黏膜念珠菌感染[6-7]。也有報道此型可能合并生長激素缺乏[8-9]。
3.3 APSⅢ型
APSⅢ型是指AITD伴有一個或多個自身免疫性疾病,但是不伴有Addison病和(或)HP。除此之外,該型的患者還可以合并性腺功能減退、淋巴細胞性垂體炎、胰島素自身免疫綜合征(Hirata病)、慢性萎縮性胃炎、惡性貧血、自身免疫性皮膚及神經肌肉疾病、系統性自身免疫性疾病等[10-11]。
3.4 APSⅣ型
這一分型目前最少見,是指兩種或兩種以上內分泌腺(器官)發生自身免疫性疾病,為AI合并其他自身免疫性疾病,但有異于Ⅰ、Ⅱ、Ⅲ型[12-16],可能合并類風濕性關節炎、原發性肝功能衰竭、乳糜瀉等,但不合并念珠菌病、AITD、T1DM。
由于APSⅠ、Ⅱ、Ⅲ型的臨床表現常有重疊,因此,近年有新的分型趨向將這三型合并為一型,單純將APS分為兩型。
綜上所述,本例患兒首先發生AITD,繼而發生T1DM,體內存在GAD、TRAb、TPOAb-R等多腺體自身免疫證據,無皮膚黏膜念珠菌感染及甲狀旁腺功能減退,故不屬于APSⅠ型;因未出現AI,故不屬于APSⅡ型;綜上所述,屬于APSⅢ型。由于APS臨床表現多樣,各種內分泌腺體功能受損的臨床癥狀可以同步也可以不同步發生,容易漏診誤診。因為各個器官病變發病時間相隔可長達20年,該患者在長期的隨訪中可能出現新的疾病,導致分型改變,故應接受長期隨訪。
[參考文獻]
[1]陳灝珠,廖履坦,楊秉輝.實用內科學[M].12版.北京:人民衛生出版社,2005:1311-1313.
[2]顏純,王慕逖.小兒內分泌學[M].2版.北京:人民衛生出版社,2006:626.
[3]Fischer A,Provot J,Jais JP,等.原發性免疫缺陷病易發生自身免疫病和炎癥表現[J].中華臨床免疫和變態反應雜志,2018,12(2):230-235.
[4]向茜,孫健瑋,馬瓊麟.自身免疫性多內分泌腺病綜合征Ⅰ型1例[J].中華骨質疏松和骨礦鹽疾病雜志,2018,11(3):284.
[5]孫永香,何亞非,栗夏連.1例自身免疫性多內分泌腺病綜合征Ⅰ型患者的臨床及家系AIRE基因突變分析[J].中國當代兒科雜志,2016,18(2):147-151.
[6]李楊,黃朱亮,萬菁菁.自身免疫性多發內分泌腺病綜合征Ⅰ型1例報道[J].國際內分泌代謝雜志,2018,38(1):63.
[7]李楊,萬菁菁,李零燕,等.自身免疫性多發內分泌腺病綜合征Ⅰ型研究進展[J].國際內分泌代謝雜志2017,37(6):426-429.
[8]Papathanasiou A,Kousta E,Skarpa V,et al.Growth hormone deficiency in a patient with autoimmune polyendocrinopathy type 2[J].Hormones,2007,6(3):247-250.
[9]謝小超,楊愛紅,劉敏,等.自身免疫性多內分泌腺病綜合征Ⅲ型1例報告[J].中國實用醫藥,2016,11(24):216-217.
[10]吳昊,李金慧,關海霞,等.自身免疫性多內分泌腺綜合征4例報告并文獻復習[J].國際內分泌代謝雜志,2016, 36(6):420-423.
[11]趙艷艷,秦貴軍.1型糖尿病與其他內分泌疾病[J].中華糖尿病雜志,2016,8(10):583-587.
[12]陳丹霞,麥鴻成,郭葉群,等.自身免疫性多內分泌腺病綜合征Ⅲ型合并胸腺增生1例報告并文獻復習[J].南昌大學學報(醫學版),2017,57(3)98-101.
[13]皮亞雷,張亞男,韓笑,等.罕見基因突變致Ⅰ型自身免疫性多內分泌腺病綜合征1例臨床及家系分析[J].臨床薈萃,2016,31(12):1318-1320.
[14]狄紅杰,陳國芳,劉超.自身免疫性多發性內分泌腺病綜合征病例報道及分子遺傳學研究進展[J].中國實用內科雜志,2018,38(10):26-29.
[15]Barker JM. Clinical.Type I diabetes associated autoimmunity:natural history,genetic associations,and screening[J].J Clin Endocrinol Metab,2006,91(4):1210-1217.
[16]李俊巖,李劍波.自身免疫性多內分泌腺病綜合征二例[J].中華糖尿病雜志,2017,9(9):581-583.
(收稿日期:2019-02-18? 本文編輯:許俊琴)
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
保健醫苑(2022年6期)2022-07-08 01:26:34
家庭科學·新健康(2022年3期)2022-05-10 00:32:13
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年11期)2021-08-22 03:15:16
中國科技論壇(2017年7期)2017-07-25 08:49:53
媽媽寶寶(2017年3期)2017-02-21 01:22:30
飼料與畜牧(規模養豬)(2016年5期)2016-12-01 03:48:40
