逆流色譜在外消旋體分離中的應用
2019-11-02 00:53:20竺少銘胡珊姍王紅衛孔望欣
浙江化工 2019年10期
竺少銘,胡珊姍,王紅衛,孔望欣
(浙江醫藥股份有限公司昌海生物分公司,浙江 紹興 312366)
0 引言
目前臨床上使用的很多藥物屬于手性藥物的消旋體,但手性藥物的不同對映體與生物大分子的作用表現不同,從而在機體內產生不同的藥理、毒理、藥代、藥效作用。服用單一對映體可以減少藥物劑量和人體代謝負擔,減少與其他藥物的相互作用,提高活性并降低由某對映體產生副作用的可能性。現有的1327 種合成藥物中含有手性中心的有528 種,接近40%,然而以外消旋體形式銷售的手性藥物幾乎高達80%。美國食品和藥物管理局(FDA)于1992 年規定,今后凡發展具有不對稱中心的藥物,必須給出手性拆分的結果。為了能準確地了解藥效和安全用藥,發展和建立簡單快速的手性藥物對映體的分離分析方法,并用于臨床研究和醫藥質量控制,已成為醫藥界關注的重大課題。獲取手性藥物單一對映體的方法有外消旋體的拆分和不對稱合成(生物合成也算作不對稱催化合成)。由于不對稱合成的合成步驟煩瑣、反應條件苛刻,外消旋體藥物或中間體的拆分成為獲取單一對映體的主要方法。外消旋體的拆分包括化學拆分、酶法拆分、色譜拆分和膜分離拆分。色譜分析技術是目前分離消旋體的主要方法之一[1-2]。采用HPLC 技術進行手性異構體的分離,需要將手性選擇試劑(CS)通過化學的手段鍵合在一種固體介質上作為HPLC 分離的固定相,這種手性分離柱的制備價格昂貴,且需要一個費時且復雜的過程;同時采用HPLC 法進行制備性分離的溶劑消耗量也很大。因此尋求高效廉價的手性分離分析新方法成為研究的熱點。
逆流色譜作為色譜技術中的一種,起源于逆流分溶法。在20 世紀80 年代隨著流體靜力學平衡系統(HSES)和流體動力學平衡系統(HDES)的提出而取得了很大的突破[3]。在HDES 基礎上,Conway W D 等在實驗室發現單側HDES 現象[3-4]。高效逆流色譜儀的出現得益于單側HDES 現象的發現[5]。1994 年Ito 博士提出了新的CCC 技術,pH 區帶逆流色譜和離子對逆流色譜[6],使逆流色譜技術有了長足發展。
高速逆流色譜不需要固體支撐體,物質的分離依據其在兩相中分配系數的不同而實現,因而避免了不可逆吸附而引起的樣品損失、失活、變性等,更能反映其本來的特性,適合于天然生物活性成分的分離,而且由于被分離物質與液態固定相之間能夠充分接觸,使得樣品的制備效率大大提高,是一種理想的制備分離手段。逆流色譜應用于手性化合物的分離,不需要采用化學手段將手性試劑鍵合到固體介質上,同一根逆流色譜分離柱可以反復多次地用于不同手性化合物的分離,只需選擇合適的兩相溶劑體系和手性試劑即可,在拆分外消旋體時避免了不可逆吸附引起的溶液和昂貴樣品的損失。相對于傳統的色譜拆分手段,高速逆流色譜具有適用范圍廣[7]、操作靈活、高效、快速、制備量大、費用低等優點。
1 逆流色譜手性分離原理
由于逆流色譜技術與一般液相色譜的主要區別是不用填料作為固定相,這與普通HPLC 相比是完全不一樣的:手性試劑不需要涂敷或鍵合到固體填料上,因此手性識別機理存在一定差異。
目前,色譜法分離外消旋體一般采用兩種形式:直接法和間接法。間接法是使對映異構體先與一種光學純的試劑反應,生成非對映異構體,然后在非手性的環境下進行分離;直接法即采用手性固定相或手性添加劑直接對異構體進行分離和測定。直接法因簡便、快捷而在色譜分離手性藥物上面應用更廣泛[8]。
逆流色譜拆分消旋體一般采用的是直接法,即在逆流色譜的固定相或者流動相中分別添加手性試劑(疏水性和親水性手性試劑的結合)來實現手性化合物的拆分和分離,主要分為單相識別模式和雙相識別(固定相Stationary phase/流動相Mobile phase Recognition,簡 稱S/M 識 別)模式。手性選擇劑和對映體之間通過氫鍵、偶極-偶極、π-π、疏水作用和空間作用等結合,從而在兩相間的分配系數不同(親和力不同),達到分離手性藥物的目的。
1.1 單相識別模式
單相識別:即僅僅在固定相中添加手性試劑或僅僅是手性流動相添加劑法,但事實上由于CCC 分離柱的理論塔板遠小于HPLC 分離柱,單一的在固定相或在流動相中添加手性試劑的方法對于CCC 來說很難取得良好的分離效果。
以有機固定相中添加手性試劑為例,高速逆流色譜手性分離單相識別原理如圖1 所示:
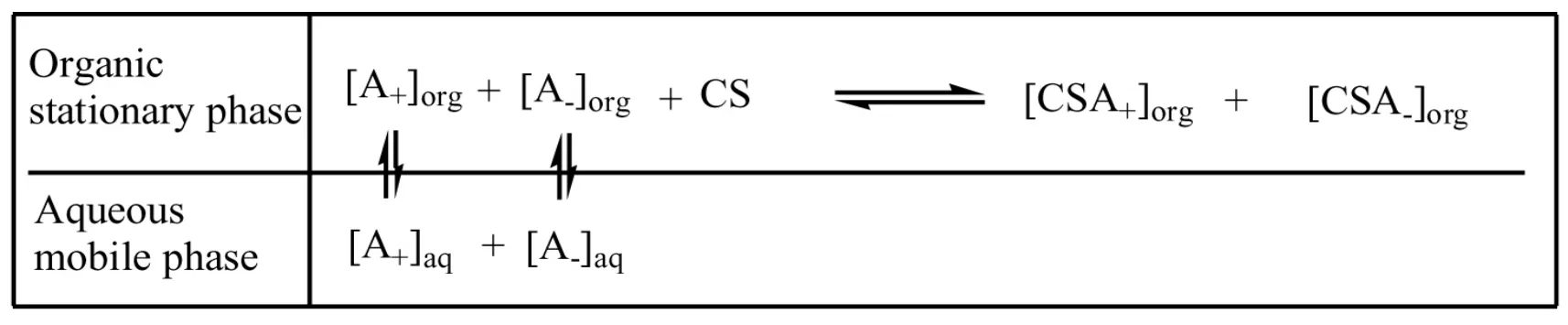
圖1 高速逆流色譜手性分離單相識別原理示意圖Fig.1 Schematic diagram of chemodynamic equilibrium between the racemates(A±)and chiral selector(CS)in the separation column based on monophasic recognition
從圖1 中可以看出,在固定相中對映體(A+和A-),手性選擇劑(CS)與它們的結合物(CSA+和CSA-)保持動態平衡。同時對映體(A+和A-)各自在固定相和流動相中保持平衡。可以得到下列表達式:

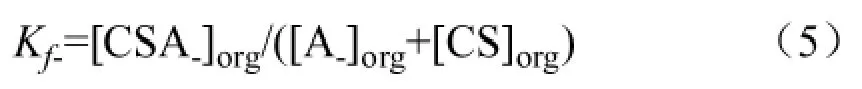
其中,A+—表示右旋體;A-—表示左旋體;A+,-—表示外消旋體;D+—表示A+在有手性添加劑的情況下的分配系數;D-—表示A-在有手性添加劑的情況下的分配系數;D0—表示A+,-在沒有手性添加劑的情況下的分配系數;Kf+—表示在化學計量比為1∶1 情況下結合物CSA+的形成常數;Kf-—表示在化學計量比為1∶1 情況下結合物CSA-的形成常數。
從上述表達式可以得到:
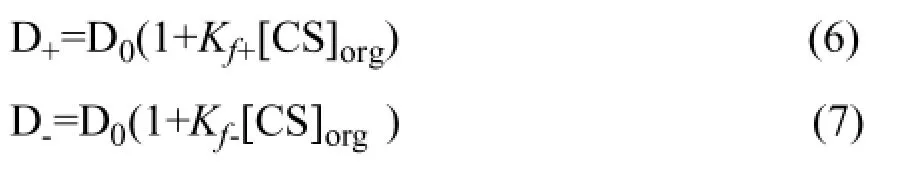
則對映體的分離因子α:
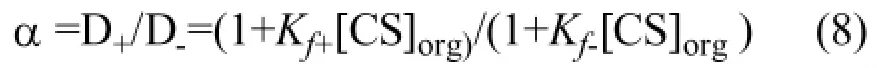
式(8)表明對映體的分離因子α 隨著手性選擇劑的濃度([CS])和Kf+/Kf-比值的增加而增加。由于與手性添加劑(CS)結合能力的不同,左旋體和右旋體在兩相體系中分配系數的差異引起分離因子變化,實現對映體分離。
事實上,由于CCC 分離柱的理論塔板遠小于HPLC 分離柱,單一的在固定相或在流動相中添加手性試劑的方法對于CCC 來說很難取得良好的分離效果。
1.2 雙相識別模式
雙相識別:即在逆流色譜的固定相和流動相中分別添加不同的手性試劑來研究手性化合物的分離。HSCCC 雙相識別原理如圖2 所示:
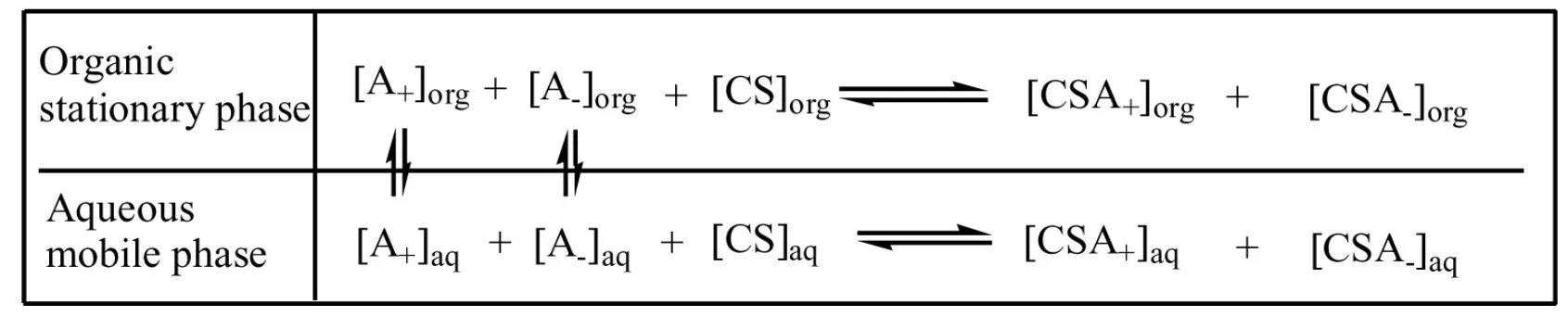
圖2 雙相識別逆流色譜原理圖Fig.2 Schematic diagram of chemodynamic equilibrium between the racemates(A±)and chiral selector(CS)in the separation column based on biphasic recognition.
從圖2 中可以看出,對映體(A+和A-),兩種不同手性選擇劑([CS]org和[CS]aq)與它們的結合物([CSA+]org、[CSA-]org和[CSA+]aq、[CSA-]aq)分別在固定相和流動相中保持動態平衡。同時對映體(A+和A-)各自在固定相和流動相中保持平衡。
相比于單相識別模式,雙相識別模式中,對映體的分配系數表示如下:
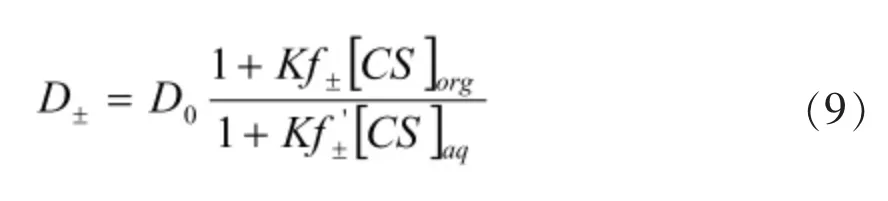
對映體的分離因子α:
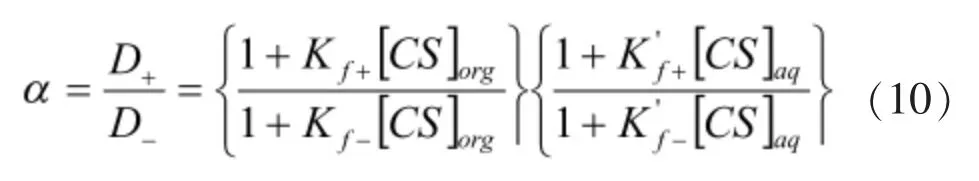
Kf+—表示在化學計量比為1∶1 情況下結合物[CSA+]org在有機相中的形成常數;Kf-—表示在化學計量比為1∶1 情況下結合物[CSA-]org在有機相中的形成常數;K′f+—表示在化學計量比為1∶1 情況下結合物[CSA+]aq在水相中的形成常數;K′f-—表示在化學計量比為1∶1 情況下結合物[CSA-]aq在水相中的形成常數。
在流動相中添加手性選擇劑,通過流動相手性選擇劑的輔助識別能力,在表觀上增大了固定相手性選擇劑對對映體的選擇性,使得分離選擇性系數增大。流動相中添加的手性選擇劑與對映體產生一種競爭吸附,能夠顯著改變外消旋體不同對映體與固定相手性選擇劑之間的分子間作用力,最終導致分離度增加。在雙相識別體系中,對映體的分離因子α 和親水性手性選擇劑的濃度([CS]aq)、親脂性手性選擇劑的濃度([CS]org)和Kf+/Kf-比值以及K′f+/K′f-比值有很大關系,隨著這些因素增加而增加。所以添加合適的手性選擇劑和選擇合適的兩相溶劑系統,對于成功分離外消旋體十分重要。
2 逆流色譜手性系統的選擇
2.1 兩相溶劑系統的選擇
溶劑體系的選擇對于HSCCC 十分重要,兩相溶劑應該滿足以下要求[9]:(1)為了保證必要的固定相保留值(體積比不小于50%),溶劑體系的分層時間應小于30 s;(2)目標樣品的分配系數K若接近于1,容量因子應大于1.5;(3)盡量采用揮發性溶劑,以方便后續處理及易于物質的純化;(4)不造成樣品的分解或變性,且對樣品有足夠高的溶解度。
目前用的最多和比較適合的是兩種溶劑系統:正己烷/乙酸乙酯/正丁醇/甲醇/水,氯仿/甲醇/水。對于一種未知的樣品,Ito 認為可以選擇一種應用較為成熟的溶劑系統,例如先采用正己烷/乙酸乙酯/甲醇/水(1∶1∶1∶1)或氯仿/甲醇/水(10∶3∶7)進行嘗試,再根據目標組分的分配系數調整溶劑的組成[10]。如果需要親水性更強的體系,加入鹽(醋酸銨)或酸(三氟醋酸或醋酸)。還有人提出正丁醇/乙酸乙酯/水(3∶2∶5)適用于分離極性大的物質,而正庚醇/乙酸乙酯/甲醇/水(6∶1∶6∶1)適用于弱極性和非極性體系[11]。
常用溶劑體系的選擇首先根據樣品的理化特性選出最佳溶劑,再選擇相應的數種溶劑,以組成選擇性最好的多元溶劑體系[12]。
2.2 添加手性選擇劑
在添加手性選擇劑時,手性選擇劑應滿足以下要求:①能夠完全溶解在溶劑中或混合溶劑中;②在液相溶劑中能夠保持對對映體的識別能力。一般地,手性選擇劑加入由有機溶劑或混合溶劑組成親脂固定相中,親水性溶劑或水溶液作為流動相。
篩選合適手性選擇劑方法:(1)從手性試劑的萃取拆分研究以及借鑒氣、液、毛細管電泳中廣泛使用的手性添加劑,找到適合于逆流色譜的手性添加劑,是一個很好的途徑,這類手性選擇劑有離子對試劑、配體交換試劑、蛋白質親和試劑、環糊精及冠醚包合試劑和手性氫鍵作用試劑等。如環糊精及其衍生物,冠醚包合試劑已經用在分離撲爾敏、氨魯米特等手性藥物逆流色譜中。(2)按照交互作用原理選擇手性選擇劑:如果化合物A 的對映體之一能夠拆分化合物B 的對映體,則化合物B 的單一對映體能夠拆分A 的對映體。目前人們嘗試以大環分子類的環糊精及其衍生物、冠醚及其衍生物,天然多糖衍生物,蛋白質和氨基酸作為手性選擇劑,在拆分消旋體方面取得了成功。其中CCC 手性分離中應用比較成功的手性選擇劑主要有三類:L-脯氨酸衍生物、磺化β-環糊精類和大環抗生素(萬古霉素)類。
2.2.1 以大環分子作為手性選擇劑
2.2.1.1 環糊精及其衍生物
組成環糊精的每個葡萄糖單元有5 個手性碳原子,因而作為主體分子,能夠提供客體分子一個良好的不對稱環境,由于獨特的結構,環糊精能選擇性地包結多種客體分子,形成具有不同自由能的包結物。作為手性選擇劑,客體分子的作用行為將呈現差別,若客體分子是一對對映體,則將形成非對映體的包結物,呈現對映體選擇性,這是環糊精拆分對映體的主要依據。
環糊精取代基的位置及引入基團的性質將深刻影響改性后的環糊精的各種性狀,成為影響其手性選擇性的主要因素[13]。研究結果表明,如果分子中的手性中心與伯氨基鄰近,可獲得最佳的分離效果[14]。環糊精外部羥基衍生化,羥基被羧甲基、甲基等基團取代,取代基具有雙重作用,在空間上擴大或縮小環糊精手性腔的大小,改變包結作用能力;同時,由于引入新的基團可發生p-p相互作用,氫鍵和偶極疊合的功能,因此,增加新的拆分作用點,兩者均導致選擇性的顯著變化。化學修飾環糊精可改變天然環糊精的理化性質,改變手性選擇性,環糊精衍生物分離手性物質的成功在HPLC,CE 和GC 有過很多報道[15-17]。
Breinholt J 等[18]采用硫酸化的β-環糊精作為選擇劑去分離7-去甲基-奧美昔芬。首次介紹了環糊精衍生物作為手性選擇劑成功應用于逆流色譜分離外消旋體。
劉佳川[19]嘗試在高速逆流色譜中以環糊精及其衍生物作為手性選擇劑,對15 種藥物進行篩選拆分,發現水溶性的羧甲基β-環糊精能夠拆分手性藥物氨魯米特和撲爾敏,并建立了制備性分離氨魯米特的分離方法。溶劑系統選擇乙酸乙酯/甲醇/水的比例為10∶1∶9 或10∶2∶8 對氨魯米特能拆分,且比例為10∶1∶9 拆分效果比較好。
Ai P 等[20]選擇20 mmol/L 羧基羧甲酯-β-環糊精作為手性選擇劑添加到流動相中,用乙酸乙酯:甲醇:水=10∶1∶9 作為兩相溶劑系統,分離1.2 h,消旋體能得到有效的分離。
Tong S Q 等[21]作了高速逆流色譜雙相識別模式分離α-環己基扁桃酸外消旋體的研究。兩相溶劑系統選擇正己烷/甲基叔丁基醚/磷酸鹽溶液(9∶1∶10),添加0.3 mol/L(-)-2-乙基己基酒石酸鹽在有機固定相中,添加0.1 mol/L 羥丙基-β-環糊精于流動相中,在溫度為8 ℃,流動相pH=2.68,樣品在帶260 mL 柱容積的CCC 制備儀中最大量為440 mg,在這種分離條件下,分離效果最好。分離物的純度可以達到99.5%以上。除掉CS 得到目標化合物,可以達到85%~88%。最終右旋對映體得到186 mg,左旋對映體得到190 mg。
2.2.1.2 冠醚及其衍生物
冠醚的結構是一種有親水性的內腔和疏水性的外殼,一般是引入一些手性單元作為手性選擇劑。目前常用的是18-冠-6-醚類,它主要用于分離一級胺,尤其是氨基酸及其衍生物的分離(醚環上的氧原子和氮原子上的氫原子形成氫鍵,4 個手性碳上的羧基向兩側交叉對應形成位阻,靜電作用和位阻作用使對映體與冠醚形成有差異的主客體)。但一級胺必須質子化方能達到分離,因此流動相必須是酸性的。
Kim E 等[22]研究應用(+)-(18-冠-6)-四甲酸作為手性選擇劑,分離吉米沙星對映體。溶劑的疏水性增加,吉米沙星單一對映體的分離度相應的增加。所以選擇了1-BuOH∕EtOAc/20 mol/L雙三羥基甲氨基醋酸鹽緩沖液,比較適合。較高的pH 與吉米沙星有很強的相互作用,也能使冠醚去質子化。選擇最優的溶劑的pH,以及合適選擇劑含量,也能提高其分離度。在pH 為6 時,吉米沙星單一對映體的分離度最高,比應用毛細管電泳(最適宜pH 為4.5)時還要好。
冠醚的優點為在一定條件下能夠對伯胺類對映體快速直接分離,無需衍生化。但冠醚的分離能力受濃度的影響大,且毒性較大,柱效相對較低,其應用受到一些限制。
2.2.2 以天然的多糖衍生物作為手性添加劑
這類物質以糖苷鍵相連,具有螺旋結構和溝槽,從而有吸附和包結的作用,并且容易獲取。衍生化能夠提供各種作用位點,容量大適合制備型色譜。目前一般是引入3,5-二硝基取代基。
Perez E 等[23]應用纖維素和直鏈淀粉的苯基酰胺(3,5-二甲基苯基氨基甲酸酯)衍生物作為逆流色譜的手性選擇劑,采用傳統洗脫方式和pH區帶逆流色譜,嘗試用幾種有機/水兩相溶劑系統分離8 種分別顯酸性、中性和堿性的目標樣品。最后發現在甲基異丁基酮(MIBK)/水溶液或甲基叔丁基醚(MTBE)/水溶液兩相體系下,對心得靜(一種血管舒張藥)和殺鼠靈兩種藥物分離較為理想。心得靜在兩相體系為甲基異丁基酮(MIBK)/水溶液,固定相為10 mol/L 二乙醇胺(DEA)以及7.5 mg/mL 纖維素衍生物,流動相為H2O(HCl 5 mol/L)的情況下,右旋對映體的純度可以達到92%,左旋對映體的純度可以達到85%。殺鼠靈在兩相體系為甲基叔丁基醚(MTBE)/水溶液兩相體系下,固定相10 mol/L 三氟乙酸(TFA)以及7.6 mg/mL 直鏈淀粉衍生物,流動相為H2O(2.5 mol/L NH4OH)的情況下,右旋對映體的純度可以達到89%,左旋對映體的純度可以達到97%。可以發現在最好的分離條件下,兩種藥物的分離度都能夠達到84%~97%。
Perez E 等[24]也作了關于使用纖維素的衍生物作為手性選擇劑應用于逆流色譜的研究,經過化學改性,纖維素作為手性選擇劑分離外消旋體有很好的效果,已經被廣泛地應用。目前多糖類衍生物作為手性選擇劑時手性識別能力較強,方法也較為成熟。
2.2.3 蛋白質和氨基酸
目前用的較多的是L-脯氨酸衍生物,一般是至少引入一個3,5-二取代基芳香環,包括有硝基、氯代或甲基基團[25]。蛋白質和對映體的作用主要為疏水和靜電作用。
Delgado B 等[26]通過比較N-十二烷酰基-L-脯氨酸-3,5-二甲基苯胺及其衍生物作為選擇劑時分離消旋體結果,發現在其結構中引入一個π電子供應基團,對其手性選擇有積極作用,但是這些改性可能降低其在親脂性溶劑中的溶解度,可能限制制備目的的適用性。L-脯氨酸衍生物作為手性選擇劑,有更強的手性選擇性。醋酸銨緩沖液保留時間較磷酸鹽緩沖液大,酸化溶劑體系,能夠使N-二硝基苯-L-亮氨酸在有機固定相中的分離度更高,從而增加被分析物的保留和分離。
Ma Y 等[27]研究應用高速逆流色譜以L-脯氨酸的衍生物N-十二烷酰基-L-脯氨酸-3,5-二甲基苯胺作為手性選擇劑對(±)-二硝基苯氨基酸消旋體進行拆分。通過比較常規和pH 區帶逆流色譜兩種拆分技術分離外消旋體的結果,發現應用pH 區帶逆流色譜分離消旋體的效果較好。使用常規技術,在2~9 h 的時間內,10 mg 到最多1 g的樣品能夠得到分離;而pH 區帶色譜中,在少于3 h 的時間內,超過2 g 的樣品得到了有效地分離。
Ma Y[28]用高速逆流色譜進行制備規模的分離手性化合物獲得了成功,并且申請了專利。在這項研究中,使用了帶有325 mL 容量柱的多層線圈行星離心儀,以N-(3,5-introbenzoyl)-D,Lamino acids 為樣品,N-十二烷酰-L-脯氨酸-3,5-二甲基苯胺作為手性選擇劑,溶劑系統選擇正己烷/乙酸乙酯/甲醇/水,對目標樣品進行了拆分。第二種操作采用pH 區帶逆流色譜,兩種異構體的特征矩形峰的重疊小于5%。
Pérez A M[29]做了關于氟化的手性選擇劑L-脯氨酸的衍生物在逆流色譜分離旋光對映體中應用的研究。在這項研究中,描述了在CCC 中應用良好、在室溫下十分穩定的溶劑系統,即一種氟化的溶劑乙氧基諾娜氟丁烷(ENFB),發現ENFB/2-PrOH/H2O(25∶35∶40)混合液較為適合分離DNB-Leu 和DNB-Leu-tBu。該研究表明,這種氟化的手性選擇劑在分離非電離手性化合物方面具有應用前景,開發了一個新的應用領域。
蛋白類作為手性選擇劑在CCC 手性分離外消旋體中應用廣泛,拆分效果也較好。但蛋白質只能與具有特定幾何構型和化學官能團的分子結合,拆分一些特定的消旋體。
2.2.4 其他
Rubio N 等[30]研究用(S)-萘普生衍生物(S)-N,N-二乙基胺萘普生作為手性選擇劑,運用多重雙模逆流色譜進行手性分離(±)-N-(3,4-順-癸基-1,2,3,4-四氫化菲-4-)-3,5-二硝基苯酰胺和N-(3,5-二硝基苯甲酰)-(±)-亮氨酸。
Franco P 等[31]研究用金雞納屬生物堿衍生物作為逆流色譜手性選擇劑分離N-氨基酸衍生物和2-芳氧基丙酸,并找到了合適的手性系統。醋酸銨緩沖液/叔戊醇/甲醇/庚烷,特別是醋酸銨緩沖液/甲基異丁基醚或二異丙基醚非常適合對消旋體進行分離。
3 展望
逆流色譜作為一種不用固態載體能實現連續有效分離的色譜技術,在手性藥物的分離分析方面有很大的應用前景。手性分離技術已經在高壓液相色譜(HPLC)、氣相色譜(GC)和毛細管電泳(CE)中得到廣泛應用,但在逆流色譜中應用很少。與其它色譜新技術相比,逆流色譜存在一些不足,在拆分外消旋體方面還有待于普及和推廣。隨著逆流技術的迅猛發展,逆流色譜在手性分離中的應用會日趨成熟。目前主要困難在于選擇適合的手性添加劑,近年來人們選用一些常用的手性選擇劑去分離手性藥物,例如冠醚被用來分離含有氨基的一類藥物,一些源自HPLC 技術的手性選擇劑如N-十二烷酰-L-脯氨酸-3,5-二甲基苯胺和β-環糊精等,應用到逆流色譜分離外消旋體中也取得很好的效果。在這些手性選擇劑中,L-脯氨酸類和環糊精類在逆流色譜分離外消旋體的應用方面比較廣泛,技術相對成熟,且這兩類成分為天然物質,低毒,價格低廉,是未來逆流色譜手性選擇劑發展方向。相信隨著研究的深入,逆流色譜會在外消旋體分離分析方面有著更加廣泛的應用前景。主要通過手性系統的研究開發,不斷發現新的手性選擇劑和溶劑系統,提高分離效率。同時也應做深入的基礎性研究,為逆流色譜手性分離技術的不斷發展和創新提供理論基礎。
