小鼠腦代謝物活體磁共振波譜分析及與離體樣本磁共振波譜與質譜定量分析的比較研究
2019-11-12 06:29:25陳煒雷和花宋濤張利民雷皓
分析化學 2019年10期
陳煒 雷和花 宋濤 張利民 雷皓

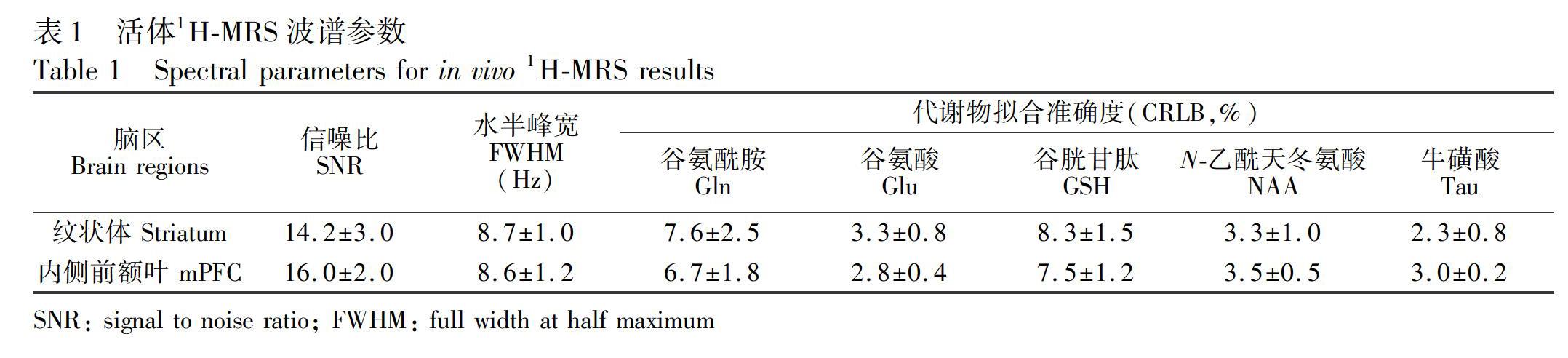
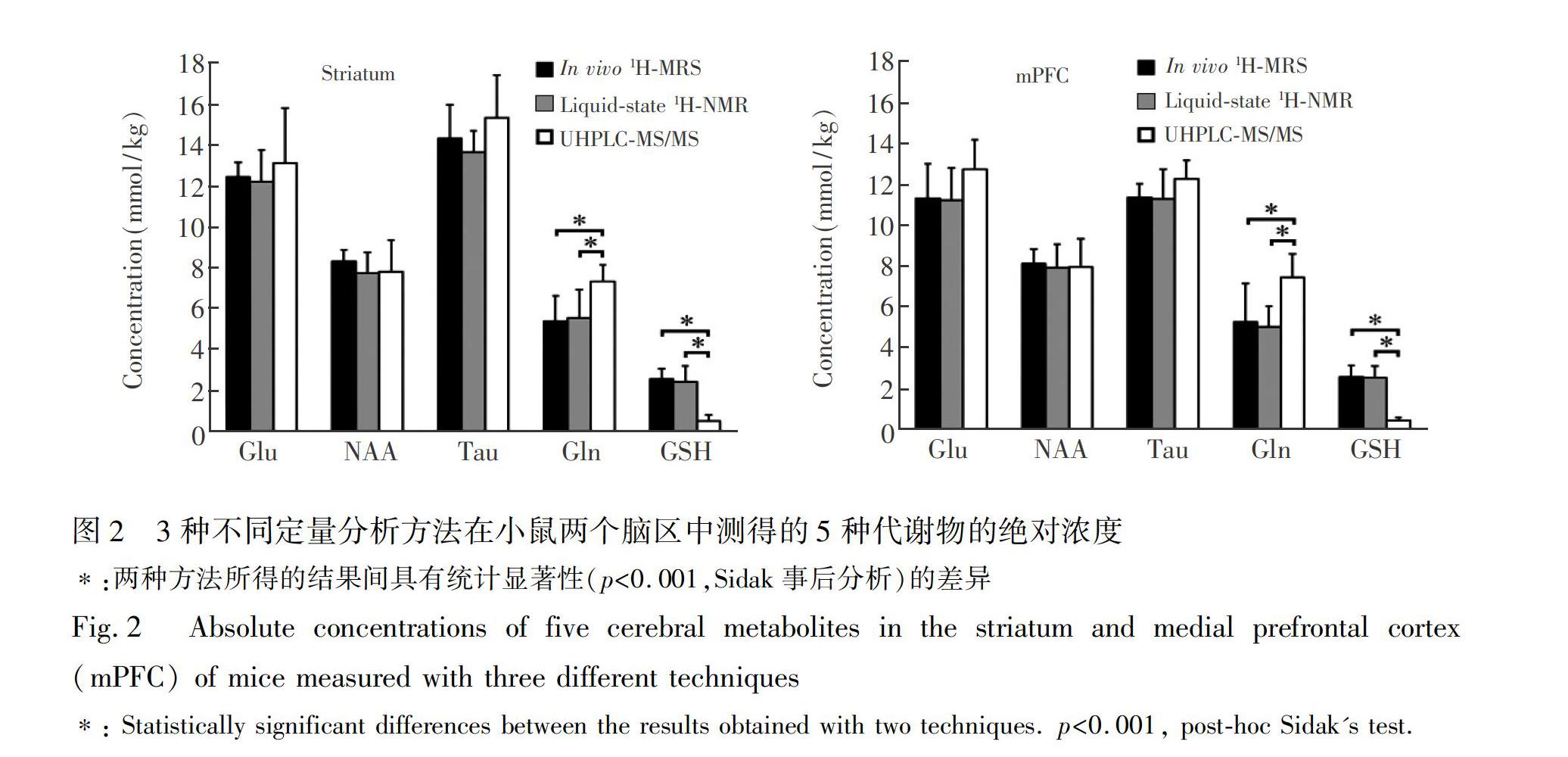
摘?要?以水為內標的活體質子磁共振波譜(1H?MRS)可非侵入性、原位、同時、定量分析多種腦代謝物的濃度,在臨床神經/精神疾病診斷、療效評估及相關基礎研究中得到廣泛應用。為驗證以水為內標的活體1H?MRS定量分析的準確性,本研究采集了小鼠紋狀體和內側前額葉腦區的活體1H?MRS,測定了N?乙酰基天冬氨酸(NAA)、谷氨酸(Glu)、牛磺酸(Tau)、谷氨酰胺(Gln)和谷胱甘肽(GSH)這5種代謝物的絕對濃度;隨后采集對應腦區的組織樣本,經萃取后用液體核磁共振波譜(1H?NMR)和超高效液相色譜?串聯質譜聯用(UHPLC?MS/MS)法定量分析上述代謝物。經統計比較發現:3種方法所測得的NAA、Glu和Tau的絕對濃度無顯著差異,且與文獻報道一致,提示活體1H?MRS定量分析具有與離體分析基本一致的準確性。活體1H?MRS和腦組織萃取樣本1H?NMR定量分析所得的Gln和GSH的絕對濃度無顯著差異。UHPLC?MS/MS測得的Gln和GSH濃度與磁共振方法所得的結果顯著不同,這可能是在樣品前處理、離子化或定量過程中引入了系統誤差所致。本研究初步驗證了聯合運用活體1H?MRS和離體磁共振/質譜方法對同一腦區中多種代謝物進行同步定量分析的可行性。
關鍵詞?質子磁共振波譜; 活體分析; 超高效液相色譜?串聯質譜; 絕對定量; 內標準
1?引 言
神經化學物質是大腦對機體生理活動進行精準調控的物質基礎[1]。神經/精神疾病的發生與發展一般都伴隨有神經化學物質的代謝紊亂[2,3]。活體、原位、定量分析神經化學物質(或腦代謝物)的濃度并動態監測其變化軌跡,對于神經/精神疾病研究、診斷和療效評估具有重要的意義。活體(In vivo)質子磁共振波譜(1H?MRS)是現有的為數不多的可無創定量檢測區域性腦代謝物濃度的方法之一,已被廣泛應用于神經/精神疾病(如帕金森氏綜合征,PD)的臨床診斷和基礎研究中[4,5]。利用活體1H?MRS技術測得的腦代謝物的濃度也被證實與其所在腦區的結構、功能或病理狀態有關[6]。
由于總肌酸(Total creatine, tCr)在正常生理狀態下一般波動較小,活體1H?MRS一般都選用其作為內標進行定量分析[7]。但在某些病理狀態下, tCr的絕對濃度也會出現顯著變化[8,9],從而影響定量分析結果。也有研究者采用水的信號為內標進行定量分析[10~12]。但這種方法也存在一些潛在的問題。 首先,腦組織中水的含量和弛豫時間(Relaxation time)在不同個體、不同發育階段及不同腦區都可能存在差異[13~15],從而給絕對定量分析帶來誤差; 其次,腦代謝物的弛豫時間各不相同,且顯著不同于水的弛豫時間,這在比較不同代謝物濃度時可能引入系統誤差[16]。
為驗證以水為內標的活體1H?MRS定量分析的準確性和可信度,本研究測定了12月齡DJ?1基因敲除小鼠2個腦區中5種代謝物的絕對濃度,并與對應腦組織萃取樣本的液體1H?NMR和超高效液相色譜?串聯質譜聯用(UHPLC?MS/MS)方法的定量分析結果進行了對比。液體1H?NMR用外加已知濃度的3?三甲基硅烷基?2,2,3,3?氘代丙酸鈉(TSP)作為內標,UHPLC?MS/MS的定量分析采用外標法,即配制一系列濃度梯度的標準溶液,建立代謝物質譜信號響應值與絕對濃度之間的標準曲線。通過比較不同方法定量分析結果的異同,驗證了聯合使用活體1H?MRS和離體方法對同一腦區中多種代謝物進行同步定量分析的可行性。
2?實驗部分
2.1?儀器與試劑
Bruker 7 T/20 cm Biospec成像儀與AVANCE Ⅲ 500核磁波譜儀(德國Bruker公司); Agilent 1290型超高效液相色譜儀串聯Agilent 6460型三重四級桿質譜儀(美國 Agilent公司); ME204/02型電子分析天平、pH計 FE28型(瑞士Mettler Toledo公司); 組織破碎儀(德國Qiagen公司); 低溫離心機(德國Eppendorf公司); Speed Vac 真空離心濃縮儀(美國Thermo公司)。
色譜純CH3CN、HCOOH、CH3OH、NAA、Glu、Gln、GSH、Tau及重水(D2O,99.9%氘代,含0.05% TSP)均購于Sigma?Aldrich公司。實驗用水為超純水(電阻率大于18.2 MΩ·cm, 由德國Merck公司 Millipore Elix Advantage 系統制備)。分析純NaCl、KCl、KH2PO4和Na2HPO4·12H2O均購于上海國藥集團化學試劑公司。
2.2?實驗動物
DJ?1基因敲除小鼠8只(由北京大學動物中心提供的種鼠繁育),體重(25.0±2.1) g,置于普通小鼠籠內,每籠2~3只,在SPF級動物房中飼養至12月齡。所有動物實驗操作均遵從中國科學院武漢物理與數學研究所的實驗動物倫理規范。采集活體1H?MRS時,小鼠用1.0%~1.5%的異氟烷?氧氣混合氣體麻醉,體溫維持在(37SymbolqB@1)℃。活體1H?MRS實驗完成后,立即斷頭,剝離大腦,放入液氮中冷凍30 s; 然后切取內側前額葉(mPFC)和紋狀體(Striatum)腦區組織,置于80℃保存。整個取樣過程在3 min內完成。提取的腦組織被均分為2份,分別用于液體1H?NMR和UHPLC?MS/MS實驗。
2.3?活體1H?MRS實驗
采用Bruker 7 T/20 cm Biospec動物磁共振成像儀,直徑為72 mm的射頻發射體線圈,單通道正交表面接收線圈。使用弛豫增強快速采集(RARE)序列采集定位像。實驗參數如下:重復時間(TR)2500 ms, 回波時間(TE)36 ms,回波鏈長(RARE factor)4,視野(FOV)20 mm×20 mm,矩陣大小128×128,片厚0.5 mm,片數20,重復次數4。活體1H?MRS采用單體元譜(PRESS)序列。實驗參數如下:TR 4000 ms,TE 15 ms,譜寬4000 Hz,采樣點數2048,帶寬為4000/3000 Hz的Hermit激發/重聚脈沖,激發頻率中心偏離水的共振頻率
709 Hz。散相梯度場強度為25 mT/m,持續時間1.5 ms,所對應的擴散加權值為0.0178 s/mm2。 采用帶寬為13500 Hz的90°雙曲正割(Sech)射頻脈沖進行體素外信號抑制。勻場后體素內水信號的半高寬(FWHM)低于10 Hz。每個腦區先采集一個重復次數為1的未壓水的譜,然后采集一個重復次數為512的壓水后的譜。壓水采用弛豫優化的變間隔射頻脈沖技術[17]。
2.4?腦組織萃取樣本的1H?NMR實驗
每只動物取約3 mg的 mPFC組織和5 mg的紋狀體組織。加入0.6 mL甲醇?水(2∶1, V/V),經組織破碎儀(20 Hz,90 s)和冰浴超聲(超聲1 min,停1 min,重復3次)破碎處理; 離心10 min(4℃,12000 r/min),收集上清液; 重復上述步驟2次并合并上清液。上清液用真空離心濃縮儀除去甲醇,冷凍干燥機除去水分。在所得的凍干粉中加入600 μL磷酸鹽重水緩沖液(0.1 mol/L,pH=7.4,0.001% (w/V) TSP,>95% D2O),4℃下12000 r/min離心10 min,取500 μL上清液置于核磁管中。
液體1H?NMR分析在配有超低溫BBO探頭的Bruker AV Ⅲ 500 MHz核磁共振波譜儀上進行,采樣脈沖序列為ZGPR。弛豫等待時間(RD)和壓水預飽和脈沖時間分別為17和2 s,采樣譜寬20 ppm,采樣時間4 s,累加次數128。樣品溫度保持在298 K。采用1H?13C HSQC二維譜進行質子信號歸屬。F2維(1H)與F1維(13C)分別采集2048與200個數據點。累加400次,F2維譜寬12 ppm,F1維譜寬175 ppm,壓水預飽和脈沖時間1.5 s,garp組合脈沖去耦。
2.5?UHPLC?MS/MS實驗
每只動物取5 mg左右的mPFC與紋狀體組織,加入0.4 mL冷甲醇(20℃冷凍12 h)、50 μL乙腈?水(1∶1,V/V,含0.1% 甲酸)及10 μL內標溶液(氘代乙酰膽堿)后,在組織破碎儀上破碎勻漿(20 Hz,90 s)。于20℃靜止30 min后,在4℃以10000 r/min離心10 min,取上清液離心濃縮揮干。所得粉末加入100 μL乙腈?水(1∶1, V/V),經尼龍66針式過濾頭(φ 13 mm,0.22 μL)過濾至色譜樣品瓶。
色譜?質譜分析采用Agilent 1290型超高效液相色譜儀串聯Agilent 6460型三重四極桿質譜儀。色譜柱為Waters ACQUITY UPLC BEH Amide column(100 mm×2.1 mm,1.7 μm)。柱溫40℃,流速0.5 mL/min, 進樣量1 μL。流動相A為乙腈?水(1∶1, V/V); 流動相B為乙腈?水(90∶10, V/V); 流動相A和B均含0.1%甲酸和10 mmol/L甲酸銨。采用線性洗脫梯度: ?0 min,100% B; 第5 min開始,逐漸切換為 100% A,平衡時間為3 min。質譜儀離子源鞘氣與干燥氣溫度均為350℃,流量均為10 L/min。 正離子檢測模式,毛細管電壓4000 V,噴嘴電壓500 V。標準曲線法進行定量分析,即用5種目標代謝物的標準品配制一系列濃度梯度的混合溶液,在優化后的質譜條件下做標準曲線。
2.6?數據處理與統計
活體1H?MRS數據采用LCModel 6.3?1A軟件[18](Stephen Provencher Inc.,奧克維爾,加拿大)處理。經相位、基線及渦流矯正等預處理后,采用分峰擬合的方法對各代謝物進行定量分析。未被壓制的水的信號作為內標,其絕對濃度設為43300 mmol/kg。當代謝物的擬合不確定性(Cramer?Rao lower bounds,CRLB)低于20%時,所得到的數據才被認為可靠并被用于最后的分析。液體1H?NMR數據采用Bruker譜儀自帶的TopSpin 3.6.1軟件處理。原始信號先乘以線寬因子為0.3 Hz的指數窗函數后進行傅里葉變換。用TSP信號(δ=0.00 ppm)定標化學位移、手動基線及相位矯正。根據積分面積計算代謝物濃度。質譜數據用 Mass Hunter Qualitative software和 Mass Hunter Quantitative software(Agilent, B.06.00)軟件處理。本研究中代謝物濃度均為平均值±標準偏差。Kolmogorov?Smirnov檢驗用于確認濃度數據分布的正態性。統計比較采用以分析方法和腦區作為獨立變量的雙因素方差分析(Two?way ANOVA)。Sidak事后檢驗評價不同分析方法所得的代謝物濃度的差異。
4?結 論
本研究利用以水為內標的活體1H?MRS定量測量了12月齡DJ?1基因敲除小鼠紋狀體和mPFC這兩個腦區中5種代謝物的絕對濃度,并將活體分析的結果與對應腦組織萃取樣本的1H?NMR和UHPLC?MS/MS定量分析的結果進行了對比。實驗結果驗證了以水為內標的活體1H?MRS定量分析的準確性,同時也初步驗證了聯合使用活體和離體方法對動物腦區進行代謝物表型的可行性。
References
1?Duarte J M N, Lei H, Mlynárik V, Gruetter R. Neuroimage, 2012, 61(2): 342-362
2?Mitolo M, Stanzani?Maserati M, Capellari S, Testa C, Rucci P, Poda R, Oppi F, Gallassi R, Sambati L, Rizzo G, Parchi P, Evangelisti S, Talozzi L, Tonon C, Lodi R, Liguori R. Neuroimage Clin., 2019, ?23: 101843
3?Mohajeri S, Bezabeh T, Ijare O B, King S B, Thomas M A, Minuk G, Lipschitz J, Kirkpatrick I, Micflikier A B, Summers R, Smith I C P. NMR Biomed., 2019, ?32(5): e4065
4?Guan J, Rong Y, Wen Y, Wu H, Qin H, Zhang Q, Chen W. Brain Behav., 2017, ?7(9): e00792
5?Pepin J, Francelle L, Carrillo?De Sauvage M A, De Longprez L, Gipchtein P, Cambon K, Valette J, Brouillet E, Flament J. Neuroimage, 2016, ?139: 53-64
6?Zhang H, Zou Y, Lei H. NMR Biomed., 2019, ?32(1): e4024
7?Li B S Y, Wang H, Gonen O. Magn. Reson. Imaging, 2003, ?21(8): 923-928
8?Goldenberg J M, Pagel M D. NMR Biomed., 2018, ?e3943
9?Carlson H L, Macmaster F P, Harris A D, Kirton A. Hum. Brain Mapp., 2017, ?38(3): 1574-1587
10?Christiansen P, Henriksen O, Stubgaard M, Gideon P, Larsson H B W. Magn. Reson. Imaging, 1993, ?11(1): 107-118
11?Soher B J, Hurd R E, Sailasuta N, Barker P B. Magn. Reson. Med., ?1996, ?36(3): 335-339
12?Brief E E, Moll R, Li D K B, Mackay A L. NMR Biomed., 2009, ?22(3): 349-354
13?Morgan J J, Kleven G A, Tulbert C D, Olson J, Horita D A, Ronca A E. NMR Biomed., 2013, ?26(6): 683-691
14?Zhang X, Liu H, Wu J, Zhang X, Liu M, Wang Y. Neurochem. Int., 2009, ?54(8): 481-487
15?Durani L W, Hamezah H S, Ibrahim N F, Yanagisawa D, Makpol S, Damanhuri H A, Tooyama I. Biochem. Biophys. Res. Commun., 2017, ?493(3): 1356-1363
16?Yamamoto T, Isobe T, Akutsu H, Masumoto T, Ando H, Sato E, Takada K, Anno I, Matsumura A. Magn. Reson. Imaging, 2015, ?33(5): 644-648
17?Thac I, Starcuk Z, Choi I Y, Gruetter R. ?Magn. Reson. Med., ?1999, ?41(4): 649-656
18?Provencher S W. NMR Biomed., 2001, ?14(4): 260-264
19?Tkac I, Dubinsky J M, Keene C D, Gruetter R, Low W C. J. Neurochem., 2007, ?100(5): 1397-1406
