幾條熱點DNA條形碼序列對海南大戟科植物鑒定能力比較
2019-11-19 08:50:32徐臘紅陸籽豪唐歷波
廣東農業科學 2019年9期
徐臘紅,陸籽豪,唐歷波,李 櫟
(1.德克薩斯農工大學,德克薩斯 卡城 77843-3741;2.海南醫學院高等職業教育學院,海南 海口 571199;3.海南醫學院基礎醫學與生命科學學院,海南 海口 571199)
【研究意義】 DNA條形碼技術是利用基因組中一段公認的標準短序列來進行物種鑒定的分子生物學技術。DNA 條形碼鑒定與其他分子鑒定方法比較具有三大優勢:鑒定結果具有良好的重復性;鑒定方法通用性強;并可通過構建數據統一的鑒定平臺,而易于推廣和標準化[1-3]。近年來,隨著對DNA 條形碼技術的深入研究人們發現該技術在藥用植物鑒定中具有廣泛的應用前景,將是中藥鑒定方法的革命性突破。目前,中藥材DNA條形碼分子鑒定指導原則建立了以 ITS2為核心、psbA-trnH為輔的植物類藥材DNA條形碼鑒定體系和以COI為主、ITS2為輔的動物類藥材DNA條形碼鑒定體系[4]。前者已納入到2010版《中國藥典》(增補本)。
【前人研究進展】 自2003年DNA條形碼被提出以來,該技術已成為全球生物分類學的研究熱點和方向。2003年,HEBERT 等對動物界11個門13 320個物種的CO1(cytochrome c oxidase subunit 1)基因序列進行分析,發現該序列種間變異能較好地區分除刺胞動物門以外的物種。隨后的研究中,COI基因被公認為動物界中標準的DNA 條形碼基因。植物由于在進化歷程中容易發生雜交、網狀進化等事件,而且同一基因在不同類群的進化速率可能不同,導致植物條形碼的研究比動物要復雜得多。近幾年提出了一些植物DNA條形碼候選序列及其組合,但尚未達成一致意見。2009年,生命條形碼聯盟植物工作組(the plant working group of the consortium for thebarcode of life,CBOL)分析了550個物種907個樣品后,推薦以rbcL+matK組成的復合序列作為整個植物界的DNA條形碼序列的通用條形碼序列,但由于擴增效率、鑒定成功率等問題,其在植物界的通用性仍受到質疑。此外,ITS2 片段在物種水平的變異較快,有更多的突變位點以區分不同的物種,因此在DNA條形碼鑒定物種方面具有潛在的研究價值。
【本研究切入點】 黎族醫藥是黎族人民在長期與疾病斗爭的醫療實踐中不斷積累的寶貴經驗的總結,是中華民族傳統醫藥的重要組成部分,由于黎族沒有自己的文字,藥物的名稱和使用方法是靠語言代代相傳,隨著現代醫藥的普及,這一寶貴的歷史醫藥文化遺產面臨失傳的危機,亟待我們進行搶救性發掘和保護[5]。據文獻記載和民間調查,五指山區黎族使用的藥用植物約有515種,隸屬于125科360屬[6-7],中大戟科植物多數種類有毒,有毒藥用植物對治療一些疾病有特殊療效,具有不可替代性,但如果鑒定錯誤很容易引起中毒事件,重者甚至會危及患者生命,為確保用藥安全,尋求準確快速的鑒定大戟科藥用植物的方法是十分必要的。
本課題組之前的研究顯示:候選DNA條形碼ITS2序列對海南大戟科植物具有較強的鑒定能力[8],但是,其他熱點DNA條形碼序列對海南大戟科植物鑒定能力如何還需進一步認證。【擬解決的關鍵問題】 因此,本研究對ITS2、ITS、psbA-trnH、rbcL、matKDNA條形碼序列對海南大戟科植物鑒定能力進行比較,以期篩選出用于鑒定海南大戟科植物最優的DNA條形碼序列,從而為建立黎族藥用大戟科植快速鑒定方法,確保用藥安全及黎藥的傳承奠定基礎。
1 材料與方法
1.1 試驗材料
供試大戟科植物材料共29個種69個樣本。樣本形態學鑒定由海南醫學院唐歷波教授完成,憑證標本及數字影像信息保存于海南醫學院基礎醫學與生命科學學院。樣本來源見表1。
1.2 試驗方法
1.2.1 植物葉片采集 每株采集健康無病斑葉片,用酒精棉球擦拭去除細菌等雜物,放入加有變色硅膠(青島海洋化工有限公司)的密封袋中進行干燥。
1.2.2 DNA提取 利用MM300 DNA球磨儀(Retsch,德國)研磨30 s(2 000 r/min)。采用植物基因組DNA提取試劑盒(天根生化科技有限公司,中國)提取總DNA。利用核酸蛋白檢測儀測定樣本DNA的濃度,然后經1%的瓊脂糖凝膠電泳進一步分析DNA提取結果。

表1 樣本來源Table 1 Source of samples
1.2.3 PCR擴增及產物測序 引物與反應條件[9-13]:本研究引物的合成由上海生工生物工程股份有限公司完成,所使用引物及其相應的反應條件見表2。PCR反應體系為25 μL,體系包括MgCl22 μL(25 mmol/L),dNTP 2 μL(2.5 mmol/L),引物各1.0 μL(2.5 μmol/L),PCR緩沖液2.5 μL,聚合酶1.0 U(博彩生物科技有限公司,中國),總DNA 1 μL(約30 ng)。PCR儀型號:promega M7501 PCR Master Mix 10 reactions TAQ。瓊脂糖凝膠電泳檢測PCR擴增結果。PCR擴增產物的測序工作由深圳華大基因生物醫學工程有限公司完成。
1.2.4 序列分析 采用PCR擴增效率、測序成功率和序列獲得率來評價5對候選序列的獲得情況。測序峰圖文件中,ITS2序列依照KELLER等[14]的方法處理,psbA-trnH序列根據GenBank上的注釋處理,其余序列采用CodonCode Aligner處理,剔除不確定堿基數目大于10個和長度小于100 bp的序列。將拼接后的序列用軟件MEGA6.0進行分析比對[15],并基于K2P 模型進行遺傳距離等分析,使用TAXON DNA軟件分析序列種內、種間變異并作barcoding gap圖。
2 結果與分析
2.1 PCR擴增率、測序成功率及序列獲得率分析 本研究通過比較5條候選序列的PCR擴增率(出現明顯PCR條帶即判定為成功)、測序成功率(測序后獲得高質量的序列即判定為成功)和序列獲得率(序列獲得率=PCR擴增效率×測序成功率)發現,ITS序列擴增效率最高、達98.4%,psbA-trnH序列居其次、為95.3%,matK序列最低、為62.5%;而測序成功率以psbA-trnH序列最高、為94.29%,其次是ITS2、為91.34%,ITS序列測序成功率最低、為78.06%;5條候選序列的序列獲得率分別為86.96%、76.81%、89.86%、79.71%、49.28%(圖1)。

表2 植物DNA條形碼通用引物序列及PCR反應條件Table 2 General primer sequence and PCR reaction conditions of plant DNA barcode

圖1 5條序列PCR擴增率、測序成功率及序列獲得率Fig.1 PCR amplification rate,sequencing success rate and sequence acquisition rate of five sequences
2.2 序列長度和變異系數分析
用MEGA 6.0軟件對所得序列進行比對分析,比較不同序列比對長度、變異位點數、信息位點數等參數。分析結果(表3)表明,ITS2序列的變異系數和信息位點系數均最大,其次為ITS序列,最小是rbcL序列;psbA-trnH序列的變異系數大于matK序列,但psbA-trnH序列的信息位點系數小于matK序列。
2.3 不同序列的Barcoding Gap分析
利用TAXON DNA軟件對不同DNA條形碼序列對供試材料種間、種內遺傳距離進行分析,并作出barcoding gap圖,結果(圖2)表明,ITS2序列有明顯的barcoding gap,種內變異、種間遺傳距離具有明顯差異,形成明顯的種間與種內變異間隔區,該序列種內變異幅度較小,種間變異幅度較大,種內變異區間為0.05~0.10,種間變異區間為0.25~0.30,種間與種內變異分布呈兩邊分開的趨勢,有利于物種鑒定;ITS序列存在barcoding gap,但不及ITS2序列顯著,種內最大變異距離值1.1716,種間最小變異距離值0.2010,且種內變異和種間變異有較多的重疊區,種內和種間變異幅度都較大,不利于物種鑒定;psbA-trnH和matK序列barcoding gap均不明顯,種內變異和種間變異有較多的重疊區,種內和種間變異幅度較大,不利于物種鑒定;rbcL序列barcoding gap不明顯,但種內變異和種間變異重疊比例較小,該序列種內變異幅度較小,種間變異幅度較大,種內最大的變異區間為0.02~0.025。不能作為單獨的條形碼鑒定物種,但可以作為ITS2的補充條形碼。
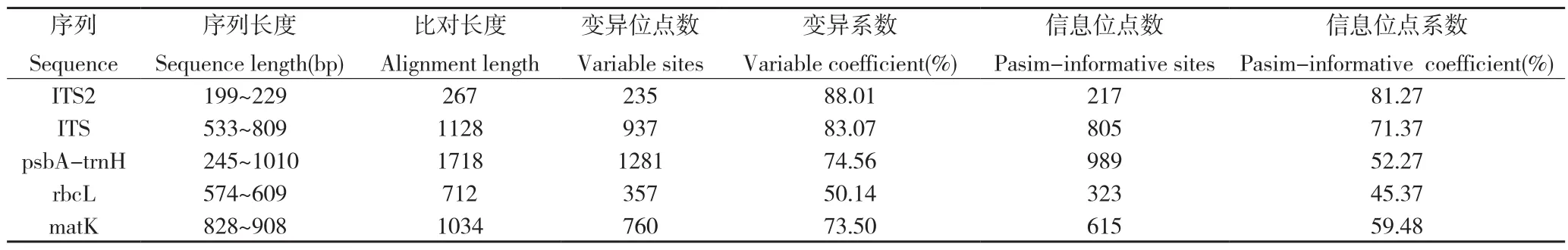
表3 5種序列的長度、變異位點和信息位點Table 3 Length,variation sites and information sites of five sequences

圖2 5個候選序列的barcoding gapFig.2 Barcoding gap of 5 candidate sequences
3 討論
DNA條形碼的核心工作之一是尋找適合的條形碼序列。 與線粒體CO1基因在動物條形碼研究中的優異表現相比,植物DNA條形碼研究進展相對緩慢[16-19]。由于植物線粒體基因組進化速率較慢,因此條形碼序列主要在葉綠體和核基因上進行選擇,被提議的候選序列主要有葉綠體編碼區rbcL、rpoC1、ycf5、matK 和非編碼區psbA-trnH序列,及核基因ITS、ITS2序列等[20-22]。
本研究通過對ITS2、ITS、psbA-trnH、rbcL、matK等熱點條形碼候選序列的分析比較發現,ITS序列種間變異要小于ITS2序列。其主要原因是ITS序列的變異主要集中在ITS1區段(平均長度為250 bp左右)和ITS2區段(平均長度250 bp左右),中間有大約160 bp的5.8 S高度保守區,因而降低了其種間變異[12],從公布的ITS通用引物來看,在各個科屬的通用性較差。本研究中,ITS序列雖然具有較高的序列獲得率,但其物種鑒定效率很低,因其具有較大的種內變異不利于分類和鑒定。
ITS2在海南大戟科物種鑒定方面具有以下優勢:首先,無論在新鮮或降解組織中都較容易擴增和測序,具有很高的應用價值;其次,ITS2序列可根據不同物種類型基于隱馬爾可夫模型的HMM 注釋方法[14]去除兩端5.8 S和26 S區段,獲得標準ITS2間隔區序列,使后續數據處理更準確可靠,這也是目前其他候選條形碼序列不具備的優勢。另外,ITS2序列具有進化速率快的特點,較高的變異程度使其在區分物種方面能力較強。psbA-trnH間隔區在被子植物中具有高度變異性[23],其兩端具有75 bp的保守區序列,便于設計通用引物[24],在大戟科中的擴增效率達95.3%,但其序列較長、存在大量的Poly(A)結構域[25],降低了測序的質量,同時在不同物種中普遍存在插入/缺失的情況,從而導致不同物種該片段長度變異較大,難以進行比對,給數據分析帶來一定的困難。從本試驗結果來看,psbA-trnH序列雖然具有較高的序列獲得率,但其物種鑒定效率很低,因其具有較大的種內變異不利于分類和鑒定。
matK序列是植物葉綠體DNA中進化較快的一條編碼區序列,目前對于matK作為DNA條形碼對植物的鑒定能力的研究仍存在較大的分歧[13,17,26]。本研究中,matK基因序列獲得率過低,會受到擴增和測序的限制,且種內與種間變異重疊比例大,物種鑒定效率很低,不適合作為大戟科DNA條形碼鑒定的候選序列。
CBOL在最新的研究中推薦rbcL+matK作為植物條形碼序列[27]。rbcL序列在GenBank中有大量的數據,并具有通用、易擴增、易比對的優點,盡管rbcL序列種內變異和種間變異barcoding gap不明顯,但種內和種間遺傳變異重疊較小,序列的種內變異幅度和變異樣本數的比例明顯較小,與其種間變異幅度和變異樣本數比例較大形成鮮明的對照,有利于區分物種。盡管rbcL基因的PCR擴增率和序列獲得率都小于ITS2序列,其在種的水平鑒定成功率也不及ITS2序列,但是可將其作為鑒別大戟科植物的DNA條形碼的補充碼,用以證明ITS2序列鑒定物種親緣關系的準確性。
4 結論
本研究通過DNA條形嗎技術,比較ITS2、ITS、psbA-trnH、rbcL、matK序列對海南大戟科植物的鑒定能力,結果表明,在以上候選DNA條形碼中,ITS2條形碼序列對海南大戟科植物鑒定能力強,可用于海南大戟科植物的快速鑒定。另外,rbcL序列不能作為單獨的條形碼鑒定物種,但可以作為ITS2的補充條形碼。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
少兒科學周刊·兒童版(2015年10期)2015-11-07 03:42:03
