乙烯與1-癸烯共聚及其共聚物微觀結構
2019-11-25 00:49:06呂春勝
石油學報(石油加工) 2019年6期
呂春勝, 成 名,, 翟 雪, 許 普
(1.東北石油大學 化學化工學院,黑龍江 大慶 163318;2.中國石油 撫順石化公司研究院,遼寧 撫順 113000)
聚烯烴在當今世界占有最大的聚合物貿易市場,2018年其產量達到165 Mt/a[1]。其中線性低密度聚乙烯(LLDPE) 是常用的塑料材料,具有環保、無毒的特性[2],應用于農業、工業和包裝行業,憑其優良的力學性能和較好的耐寒、耐候性,在國內被廣泛用于地膜和棚膜[3]。通常,LLDPE是通過Ziegler-Natta催化劑或單中心催化劑催化乙烯與α烯烴共聚獲得的[4]。LLDPE產品中,共聚物單體主要包括1-丁烯、1-己烯和1-辛烯,其中,1-己烯共聚PE是國內增長最快的PE品種[5]。聚合物的綜合性能隨著共聚單體的含碳數目增加而增強[6-7],但是在聚合反應中相同條件下隨著共聚單體的鏈增長,反應的催化活性會降低[8],因此,選擇合適的共聚單體和催化體系對開發高檔聚烯烴產品十分關鍵。1-癸烯作為一種重要的α烯烴,有廣泛應用,不僅可以作為合成潤滑油基礎油的主要原料[9-10],而且可以作為共聚單體合成LLDPE[11-12]、聚烯烴彈性體(POE)[13-15]、烯烴嵌段共聚物(OBC)[16-17]等高性能聚合材料,有較好的市場前景[18]。Wannaborworn等[12]發現,用racEt[Ind]2ZrCl2/MMAO催化乙烯與α烯烴(C6~C18)共聚制備LLDPE時,隨著共單體鏈增長,其共聚物的結晶度傾向于降低。任靜等[13]使用非茂鈦配合物制備POE時發現,與乙烯/1-辛烯共聚物相比,乙烯/1-癸烯共聚物具有更好的柔軟性和彈性。Furuyama等[17]使用Ti-FI催化劑催化乙烯與α-烯烴共聚時,所得OBC具有較好的延展性和韌性。
自Kaminsky發現用MAO活化的茂金屬催化劑可以用于烯烴聚合以來,茂金屬催化劑在聚烯烴行業中迅速發展,并由最初的雙茂型茂金屬催化劑發展到單茂型。單茂型茂金屬催化劑的典型代表為限制幾何茂金屬催化劑(CGC),其在乙烯/α-烯烴共聚反應中,具有高活性、高α-烯烴競聚率、高熱穩定性和高相對分子質量等優點[19]。然而在共聚時,其配體對共聚單體的競聚率有著較大的影響,微觀上表現為對共聚物微觀結構的影響;宏觀上則表現為對共聚物物理性能的影響[20]。因此,筆者采用2個限制幾何茂金屬催化劑分別和Al(iBu)3/[Ph3C]+[B(C6F5)4]-助劑體系,通過乙烯與1-癸烯共聚,對共聚物微觀結構的序列分布和鏈增長基元反應概率統計進行分析,探討了該體系下的共聚機理和共聚物結構,為該類催化劑配體進行有效修飾提供一定的參考。
1 實驗部分
1.1 實驗材料
催化劑合成原料:2,3,4,5-四甲基-2-環戊烯酮、正丁基鋰、四氯化鈦、三異丁基鋁,均為Aldrich公司產品。聚合反應原料:乙烯(聚合級),由中國石油大慶石油化工股份公司提供;1-癸烯(聚合級),Aldrich公司產品;甲苯(分析純),沈陽市華東試劑廠產品。
1.2 催化劑制備和聚合步驟
催化劑A、B和助劑[Ph3C]+[B(C6F5)4]-參照文獻[21-22]方法合成。其中,催化劑A為2-四甲基環戊二烯基-6-叔丁基苯氧基二氯化鈦;催化劑B為2-四甲基環戊二烯基-4,6-二叔丁基苯氧基二氯化鈦。它們的結構如圖1 所示。
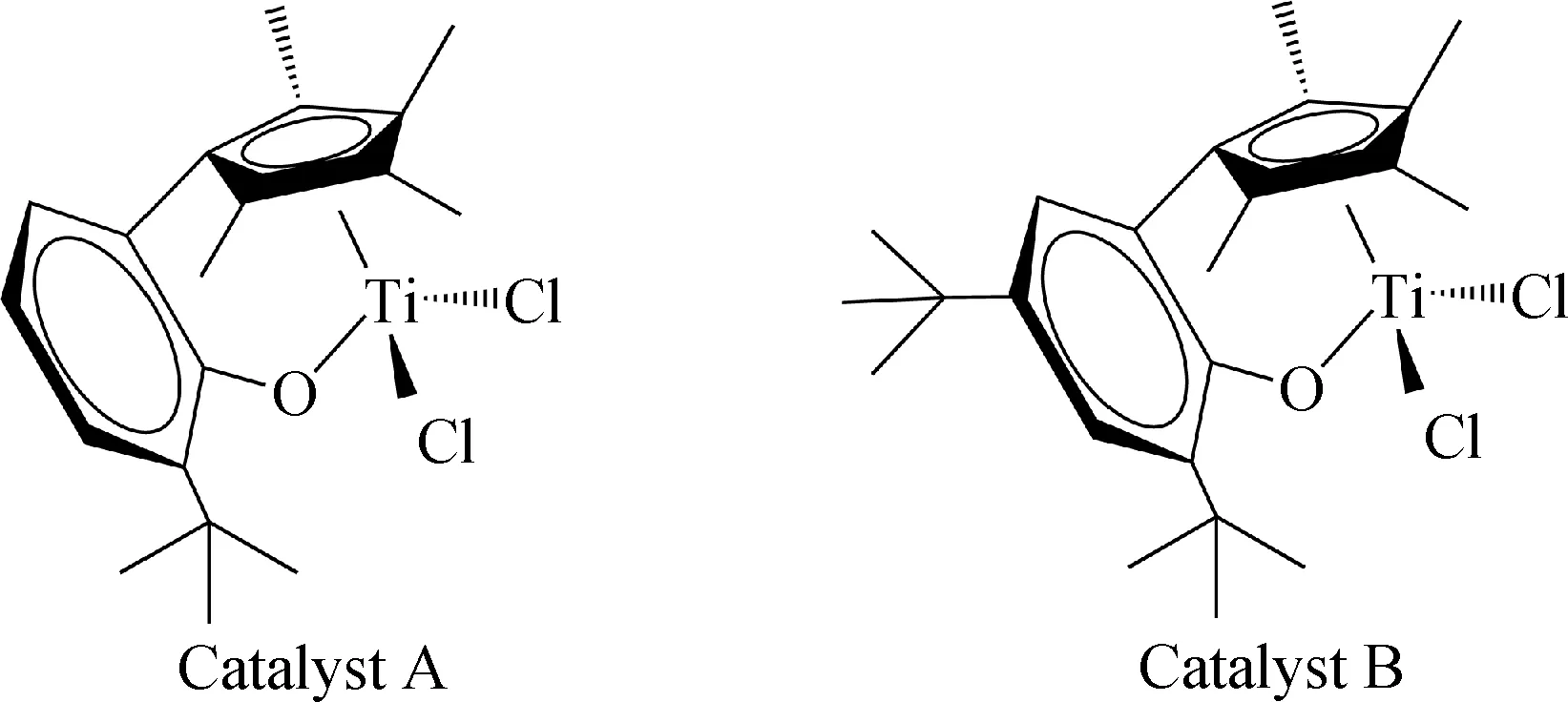
圖1 催化劑A和B的結構示意圖Fig.1 The structure of the catalyst A and B
聚合反應前,原料需要進行預處理:將乙烯通過裝有5A分子篩和MnO的精制柱進行干燥處理;將1-癸烯使用CaH2干燥處理;將甲苯在N2保護下用金屬鈉進行回流干燥。在摩爾比n(Al)/n(Ti)=100和n(B)/n(Ti)=1.2條件下,乙烯與1-癸烯共聚反應主要分4個步驟:第一步,加入原料——在無氧條件下(N2保護)將80 mL甲苯和適量共單體1-癸烯混合溶液加入至250 mL不銹鋼間歇釜(帶有磁力攪拌器)中,升溫至所需溫度,并通入乙烯(0.1 MPa)進行飽和。第二步,引發聚合——向反應釜注入主催化劑和助劑的甲苯溶液,反應壓力同時調至所需值,并通過進料氣維持壓力不變。第三步,終止反應——反應進行30 min后,向反應釜注入酸化甲醇(摩爾比為1/1的甲醇和3 mol/L鹽酸混合溶液),終止聚合。第四步,收集產物——先過濾得到聚合物,然后分別用水、甲醇洗滌,最后置于60 ℃的真空干燥箱中干燥至恒重。實驗發現,僅當加入1-癸烯濃度為0.4 mol/L時,分別由催化劑A和B催化所得的聚合物均為蠟油,其他條件得到的聚合物均為固體物。
1.3 聚合物表征
采用AV400型核磁共振儀13C-NMR分析聚合物結構,實驗條件:溶解室溫度135 ℃,共振頻率400 MHz,脈沖寬度90°,脈沖延遲時間8 s,俘獲時間1 s,樣品溶于鄰二氯苯(ODCB)中。
采用DSC 204型差示掃描量熱儀測定聚合物結晶度、熔點、熔融熱,實驗條件:樣品在N2保護下,以速率10 ℃/min從20 ℃加熱至180 ℃。
采用PL-GPC220型凝膠滲透色譜儀測定聚合物的相對分子質量(Mw)和相對分子質量分布,實驗條件:淋洗劑為1,2,4-三氯苯(TCB),測定溫度為135 ℃。
采用JSM-6360LA型掃描電鏡測定聚合物晶相結構。
2 結果與討論
2.1 乙烯與1-癸烯共聚物的物性
表1為乙烯與1-癸烯在2個限制幾何茂金屬催化劑A和B體系中進行共聚的反應條件和產物性質(共聚物的結晶度、熔融溫度、相對分子質量及其相對分子質量分布)。從表1可以得到,在相同的催化體系下,隨著共聚單體1-癸烯的初始濃度增加,乙烯與1-癸烯共聚物結晶度、熔融溫度、相對分子質量降低。這是因為,在一定范圍內,當1-癸烯濃度增加時,增加了共聚物中1-癸烯的插入率,使共聚物結晶區域被破壞程度增加,即形成的無定形區域增加,導致共聚物結晶度和熔融溫度降低[13];同時,加快了共聚反應中β-H消除速率,使共聚物的相對分子質量降低;而且,1-癸烯的插入會使乙烯序列長度降低,造成共聚物相對分子質量降低。在1-癸烯初始濃度一定時,限制型幾何茂金屬催化劑B的催化活性要高于催化劑A。這歸因于酚鹽上取代基的電子效應[23]。
圖2為1-癸烯濃度對乙烯與1-癸烯共聚物熔融曲線的影響。圖2中曲線(1)是催化劑A,共聚單體1-癸烯濃度為0.2 mol/L時所得共聚物在第二次升溫DSC曲線上得到的熔融曲線;曲線(2)、(3)、(4)則是催化劑B,共聚單體濃度分別為0.2、0.3、0.4 mol/L時所得共聚物的熔融曲線。由圖2可知,同一催化體系下,共聚物的熔融溫度隨著共聚單體1-癸烯濃度增加逐漸降低,尤其當1-癸烯濃度為0.4 mol/L時,共聚物熔融曲線基本為一條直線,說明共聚物此時呈現完全無定形態。共聚物的熔融溫度一般依賴于其結晶結構和相對分子質量。如前面所述,隨著1-癸烯濃度增加,不僅共聚物相對分子質量會降低,而且共聚物無定形區域增加,共聚物更趨近于無定形結構,因此,共聚物的熔融溫度會降低。共聚單體濃度相同時,催化劑B所得共聚物的熔融溫度高于催化劑A,正是因為取代基的電子效應,使得催化劑B的催化活性更高,所得共聚物相對分子質量更高,其熔融溫度更高。
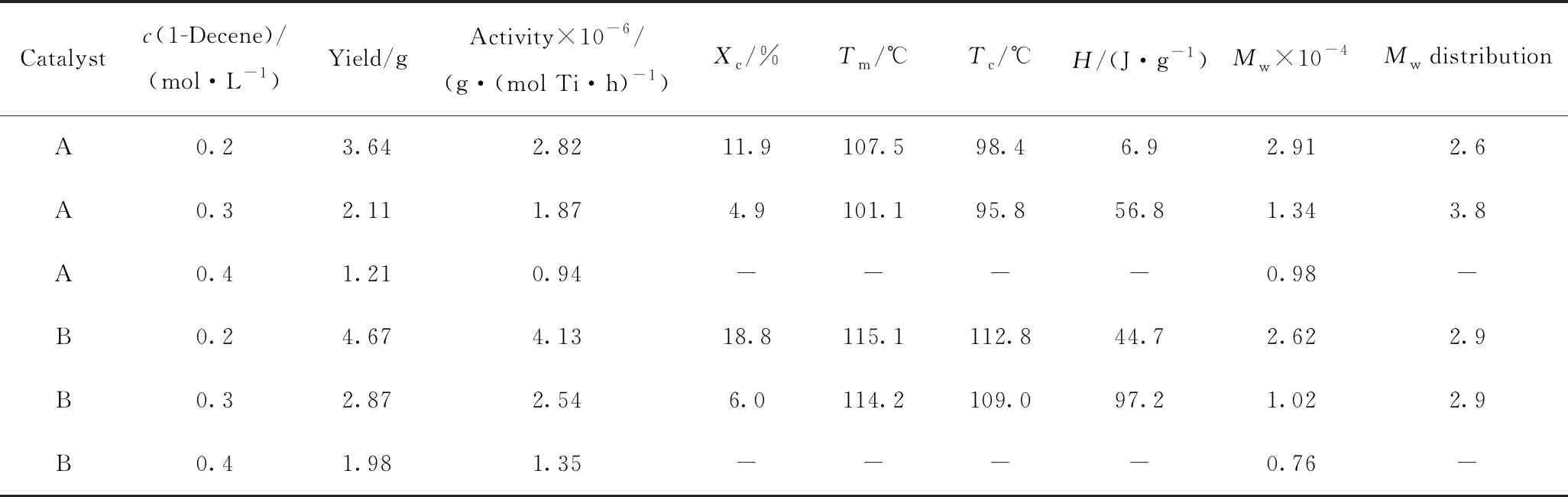
表1 乙烯和1-癸烯在催化劑A和B下的聚合條件和產物性質Table 1 Polymerization conditions of ethylene and 1-decene catalyzed by catalyst A or B and the properties of the copolymer
Polymerization conditions:T=80 ℃;p=0.5 MPa;t=30 min;m(Catalyst)=1 mg;V(Toluene)=50 mL;n(Al)/n(Ti)=100;n(B)/n(Ti)=1.2
Xc—Crystallinity of copolymer,Xc=(ΔHf/ΔHF) (100, where ΔHfis the heat of fusion of the sample as determined from the DSC curve, and ΔHFis the heat of fusion of folded-chain polyethylene (269.9 J/g)[24]);Tm—Melting temperature of copolymer;Tc—Critical temperature of copolymer;H—Enthalpy of copolymer;Mw—Relative molecular mass
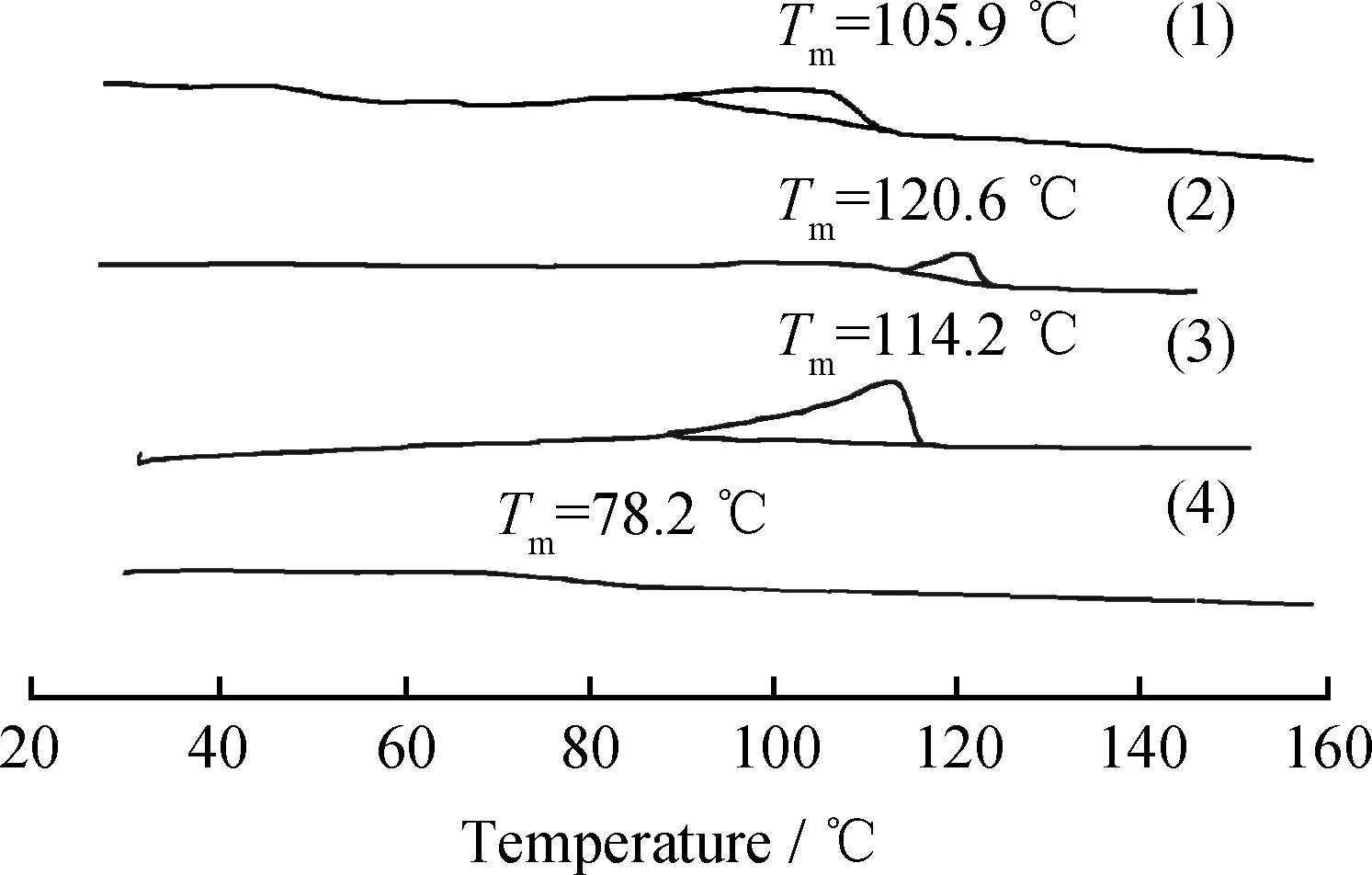
圖2 1-癸烯濃度對乙烯與1-癸烯共聚物熔融曲線的影響Fig.2 The effect of 1-decene concentrations on themelting curves of copolymer of ethylene and 1-decene(1) Catalyst A, c(1-Decene)=0.2 mol/L;(2) Catalyst B, c(1-Decene)=0.2 mol/L;(3) Catalyst B, c(1-Decene)=0.3 mol/L;(4) Catalyst B, c(1-Decene)=0.4 mol/L
2.2 乙烯與1-癸烯共聚物的微觀結構
圖3為乙烯與1-癸烯共聚物的碳標識[25]和13C NMR 譜圖。圖3(a)對聚合物鏈上的碳原子進行了標識。其中S表示聚合物主鏈上亞甲基碳原子,下標表示其從兩個方向與主鏈上相鄰叔碳原子的距離;C8表示聚合物支鏈上碳原子,1~8表示其與主鏈上相鄰叔碳原子的距離;圖3(b)中將共聚物碳核磁譜圖中的共振峰和區域劃分[26](a~h)標記在相應的位置,采用“面積歸屬法”計算出共聚物三元序列分布,依次計算二元和一元序列分布[27],其結果見表2。其中,E為乙烯鏈節,D為1-癸烯鏈節。
從表2可以得到,當催化體系一定時,隨著1-癸烯初始濃度的增加,共聚物三元序列(DDE+EDD、EED+DEE)、二元序列(ED+DE、DD)、一元序列D分布增加;相反,三元序列EEE、二元序列EE、一元序列E分布降低。其中(DDE+EDD)分布的增加會引起DD分布的增加;而EEE分布的降低造成EE分布的降低[26]。隨著1-癸烯濃度的增加,共聚物中1-癸烯含量會增加,造成rD升高、rE降低;當1-癸烯濃度為0.4 mol/L時,共聚物呈完全無定形態,而EEE分布的逐漸降低佐證了這一現象。當1-癸烯初始濃度一定時,催化劑B催化聚合共聚物的(DDE+EDD)、(EED+DEE)、(ED+DE)、DD分布高于催化劑A催化反應得到的聚合物,而EEE、EE分布趨勢相反,表明催化劑B催化更有利于1-癸烯插入共聚物主鏈上。
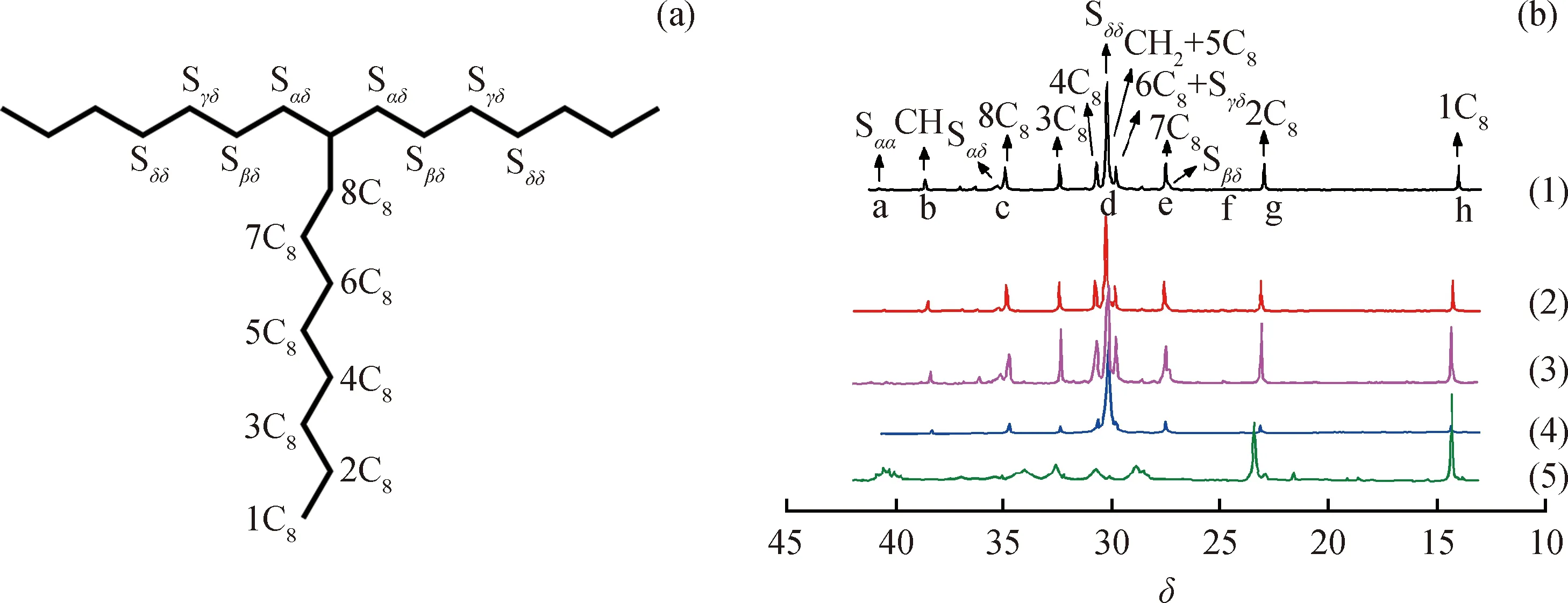
圖3 乙烯與1-癸烯共聚物的碳標識和13C NMR譜圖Fig.3 Carbon mark and 13C NMR spectra of the copolymers of ethylene and 1-decene(a) Carbon mark; (b) 13C-NMR spectra (1) Catalyst A, c(1-Decene)=0.2 mol/L; (2) Catalyst A, c(1-Decene)=0.3 mol/L;(3) Catalyst B, c(1-Decene)=0.2 mol/L; (4) Catalyst B, c(1-Decene)=0.3 mol/L; (5) Catalyst B, c(1-Decene)=0.4 mol/L
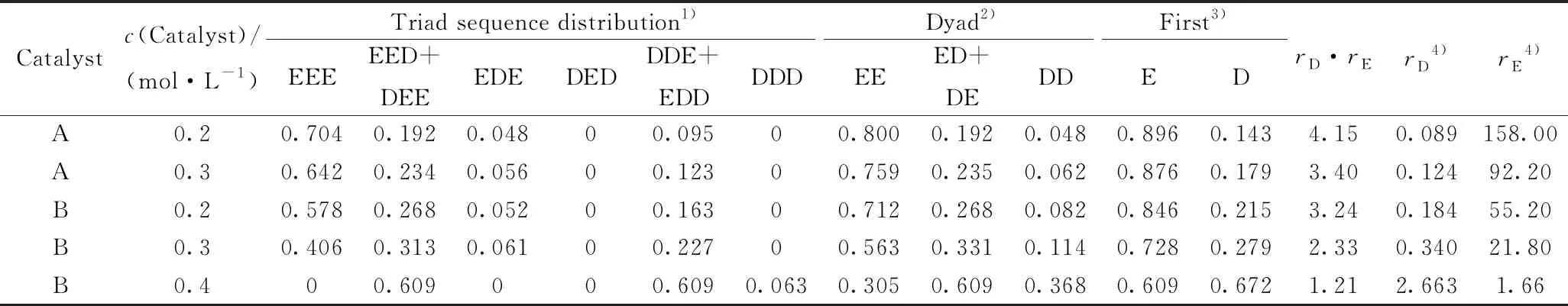
Catalystc(Catalyst)/(mol·L-1)Triad sequence distribution1)Dyad2)First3)EEEEED+DEEEDEDEDDDE+EDDDDDEEED+DEDDEDrD·rErD4)rE4)A0.20.7040.1920.04800.09500.8000.1920.0480.8960.1434.150.089158.00A0.30.6420.2340.05600.12300.7590.2350.0620.8760.1793.400.12492.20B0.20.5780.2680.05200.16300.7120.2680.0820.8460.2153.240.18455.20B0.30.4060.3130.06100.22700.5630.3310.1140.7280.2792.330.34021.80B0.400.609000.6090.0630.3050.6090.3680.6090.6721.212.6631.66
For detailed polymerization conditions, see Table 1; 1) Estimated by13C-NMR spectra; 2) EE=EEE+1/2(EED + DEE), ED=DED+EDE+1/2[(EED+DEE)+(DDE+EDD)], DD=DDD+1/2(DDE+EDD); 3) E=EE+(ED+DE)/2, D=DD+(ED+DE)/2; 4)rE·rD=4EE×DD/(ED+DE)2,rE=(2EE×E)/((ED+DE)×D),rD=(2DD×D)/((ED+DE)×E).
EEE, EED+DEE, EDE, DED, DDE+EDD, DDD—Triad sequence distribution of ethylene and/or 1-decene; EE, ED+DE, DD—Diad sequence distribution of ethylene and/ or 1-decene; E—Ethylene chain unit on copolymer chains, D—1-Decene chain unit on copolymer chains;rD—The estimated 1-decene monomer reactivity ratios,rE—The estimated ethylene monomer reactivity ratios.
表3為乙烯與1-癸烯共聚物的三元序列統計分析,其中Bernoullian模型和一級Markovian模型[28]三元序列是由表2中的實驗值二元序列計算所得。從表3可以看出,大多數共聚物的三元序列分布更符合一級Markovian模型,即聚合物增長鏈的反應性受到末端單體鏈種類的影響。這正是茂金屬催化劑在乙烯與α-烯烴共聚反應中的特點[28]。
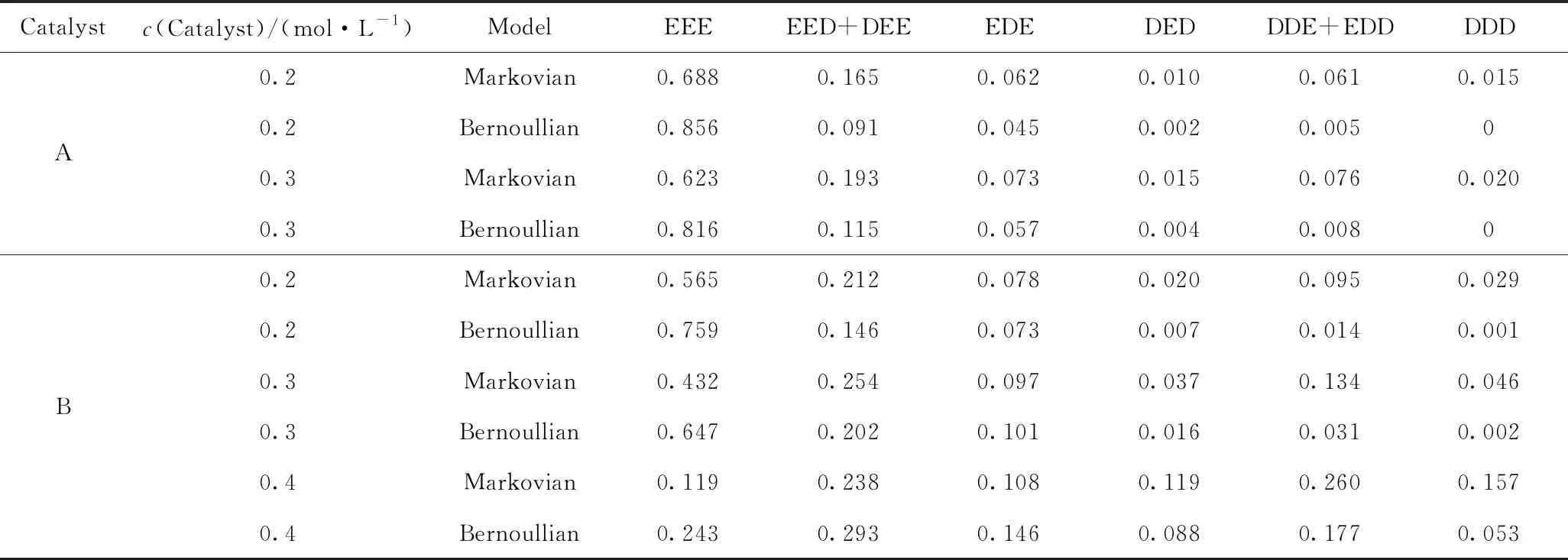
表3 乙烯與1-癸烯共聚物三元序列分布統計分析Table 3 Statistical analysis of the ternary sequence distribution of ethylene and 1-decene copolymer
表4為乙烯與1-癸烯共聚物根據一級Markovian模型所得到的概率參數。其中:PEE和PED分別指最后鏈節是E時,乙烯和1-癸烯插入時的概率。PDE和PDD分別指最后鏈節是D時,E和D插入時的概率。從表4可以看出:(1)不同條件下,大部分概率PDE>0.5、PED<0.5,表明所得共聚物結構更趨向于乙烯共聚物,而非嵌段結構和交替結構,具體結構見圖4。PDD>PED表明,與{共聚物主鏈-乙烯-1-癸烯-催化劑}序列相比,共聚單體1-癸烯要更容易配位或插入到{共聚物主鏈-1-癸烯-1癸烯-催化劑}序列;PEE>PDE表明,與{共聚物主鏈-1-癸烯-乙烯-催化劑}序列相比,乙烯更容易配位或插入到{共聚物主鏈-乙烯-乙烯-催化劑}序列。(2)同一催化體系下,隨著1-癸烯濃度的增加,PDE有變小的趨勢,PED有變大的趨勢,表明乙烯與1-癸烯共聚物隨著共聚單體1-癸烯初始濃度的增加更趨向嵌段結構(PDE<0.5、PED>0.5)。(3)催化劑B與催化劑A相比,其概率參數PDD更大,表明催化劑B體系更利于1-癸烯插入聚合物主鏈上。這是由于催化劑B與A相比,其酚基中多一個叔丁基取代基,在共聚反應中產生的電子效應更利于1-癸烯插入催化劑B中。這與Kim等[24]用Me2C(Cp)(Flu)ZrMe2和Et(Cp)(Flu)ZrMe2催化劑催化乙烯與1-癸烯共聚得到的結果一致。
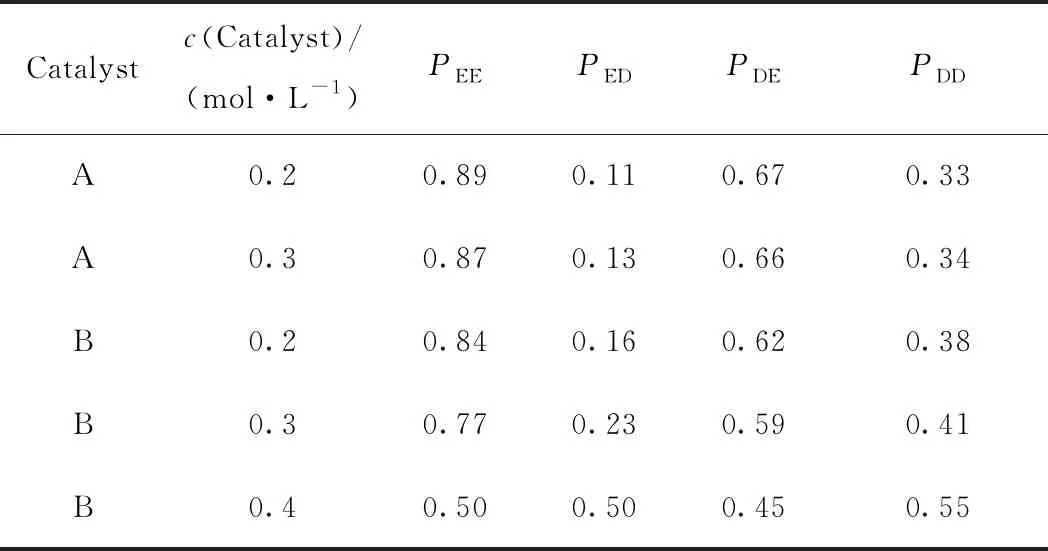
表4 乙烯與1-癸烯共聚物的一級Markovian模型概率參數Table 4 Probability parameters of copolymers of ethylene and1-decene by first order Markovian model
圖4為在c(1-Decene)=0.2 mol/L條件下采用Catalyst A所得乙烯與1-癸烯共聚物的掃描電鏡照片。由圖4可以清晰看出,共聚物形貌是纖維狀的,進一步印證所得共聚物結構趨向于聚乙烯共聚物。

圖4 乙烯與1-癸烯共聚物的掃描電鏡顯微照片Fig.4 SEM micrograph of the copolymers ofethylene and 1-decene(a) 1∶6000; (b) 1∶20000
3 結 論
采用2個限制幾何茂金屬催化劑A(2-四甲基環戊二烯基-6-叔丁基苯氧基二氯化鈦)和催化劑B(2-四甲基環戊二烯基-4,6-二叔丁基苯氧基二氯化鈦),以Al(iBu)3/[Ph3C]+[B(C6F5)4]-為助劑催化乙烯與1-癸烯共聚。得出以下結論:
(1)所得乙烯與1-癸烯共聚物Mw為(0.76~2.91)×104,Tm為101.1~115.1 ℃,共聚物主鏈上共聚單體1-癸烯質量分數為14.3%~67.2%;隨著1-癸烯濃度的增加,乙烯與1-癸烯共聚物更趨向嵌段結構(PDE<0.5、PED>0.5);1-癸烯更容易插入到催化劑B上。
(2)共聚物鏈增長機理與一級Markovian模型鏈增長機理表現相一致。
(3)乙烯利1-癸烯插入鏈節的概率參數PDE>0.5、PED<0.5,表明所得共聚物結構更趨向聚乙烯聚合物;PDD>PED表明,與{共聚物主鏈-乙烯-1-癸烯-催化劑}序列相比,共聚單體1-癸烯更容易配位或插入到{共聚物主鏈-1-癸烯-1癸烯-催化劑}序列;PEE>PDE表明,與{共聚物主鏈-1-癸烯-乙烯-催化劑}序列相比,乙烯更容易配位或插入到{共聚物主鏈-乙烯-乙烯-催化劑}序列。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
