以消化道癥狀為首發表現的家族性淀粉樣多發神經病一家系報道并文獻復習
2019-11-29 03:03:26張麗環張曉燕王賀波
疑難病雜志 2019年11期
關鍵詞:癥狀
張麗環,張曉燕,王賀波
家族性淀粉樣多發神經病(familial amyloid polyneuropathy,FAP)屬罕見的常染色體顯性遺傳性疾病,通常是由于蛋白質基因突變導致水溶性蛋白形成類淀粉纖維,在細胞外沉積而致病。相關的致病蛋白有轉甲狀腺素蛋白(transthyretin,TTR)、載脂蛋白 A-1和凝溶膠蛋白(gelsolin),其中以TTR型最為常見[1]。TTR基因的點突變是導致 TTR型FAP的主要原因,目前已在TTR基因上發現近一百多個相關的突變點位與缺損,最常見的是Val30Met(18號染色體長臂第30個氨基酸由原本的纈氨酸突變為甲硫氨酸)突變,在全球范圍內約占1/2,且分布于不同種族間[2-3]。疾病早期多以下肢感覺異常為首發癥狀,逐漸累及運動神經,伴有自主神經病變,最后死于惡病質。盡管感覺和運動表現是主要癥狀,但首發臨床表現可以是自主神經病變癥狀[4]。現對一個以消化道癥狀為首發表現的FAP家系進行分析總結,并進行相關文獻復習。
1 臨床資料
患者,女,39歲。主因“胃脹、飲食不佳5年,四肢無力3個月,加重20 d”于2018年1月31日入院。患者5年前出現胃脹、飲食不佳,伴惡心,偶有嘔吐,伴腹瀉,每日3~4次,于當地醫院就診行胃鏡檢查示慢性非萎縮性胃炎,給予中藥、護胃等藥物治療后自覺癥狀稍有改善。近2年來患者仍有胃脹、飲食不佳、腹瀉癥狀。3個月前出現活動后肢體無力,休息后緩解。后癥狀逐漸加重,日常體力活動受影響,步行1 km即感肢體無力;入院前20 d患者肢體無力癥狀明顯加重,持筷子費力,不能系扣子,不能正常蹲起,常伴頭暈發作,尤以起立時顯著,偶伴心慌,曾暈厥1次。患者自發病以來精神可,飲食、睡眠差,體質量較1年前輕約7 kg。既往體健。入院查體:T 36℃,P 79次/min,R 19次/min,BP 119/88 mmHg。發育正常,重度營養不良。心肺腹檢查未見明顯異常。專科查體:神清語利,高級皮質功能正常,顱神經檢查未見異常。雙上肢肌力Ⅳ+級,雙手拇間肌萎縮,雙下肢遠端肌力Ⅳ-級,近端肌力Ⅳ+級,雙下肢腱反射減弱,淺感覺減弱,深感覺無明顯異常,雙側Hoffmann征陰性,雙側Babinski、Chaddock 征未引出,頸無抵抗,Kernig 征陰性。
輔助檢查:甲狀腺功能六項、凝血四項、電解質、女性腫瘤全項、乙肝五項+丙抗、糞便分析、血同型半胱氨酸、血清鐵代謝、抗核抗體、抗核抗體譜未見明顯異常。生化全項:血清總蛋白62.4 g/L,白蛋白39.45 g/L。尿液分析:酮體(±),上皮細胞47.7/μl。血常規:白細胞2.09×109/L,中性粒細胞0.99×109/L,紅細胞計數3.27×1012/L,血紅蛋白98 g/L。維生素B12>2 000 pg/ml。骨髓穿刺涂片:骨髓增生較低下(約20%),粒紅比例減低,粒系各階段細胞可見,以中幼及以下階段細胞為主;紅系以中晚幼及以下紅細胞為主;巨核細胞多見,分葉核為主;淋巴細胞、漿細胞散在分布。24 h動態血壓平均值94/64 mmHg,白晝血壓平均值83/53 mmHg,夜間血壓平均值94/64 mmHg。心電圖:V1、V2導聯呈QS型,肢體導聯QRS波低電壓,V5、V6導聯電壓小于1.0 mV。24 h動態心電圖:存在短陣室速。心臟超聲心動圖示:心肌彌漫性病變(心肌淀粉樣變),二、三尖瓣少量反流,左心室舒張功能減低,少量心包積液(見圖1)。腹部超聲示:膀胱、膽囊沉積物。頭顱MR檢查未見明顯異常。胸部CT示:右肺下葉外基底段及雙肺下葉后基底段微小結節,雙肺下葉后基底段小條索,少量心包積液。神經電生理檢查:被檢測肌肉呈神經元性損害,四肢周圍神經損害(運動、感覺纖維均受累);基因檢測:患者TTR基因c.148G>T(p.V50L)雜合變異,未在受檢者母親外周血中檢出(見圖2)。其余相關家屬拒絕抽血未行基因檢測。最終診斷家族性淀粉樣多發神經病。給予注射用硫辛酸、甲鈷胺注射液營養神經,右佐匹克隆改善睡眠,地榆升白片提升白細胞,莫沙比利促進消化,胺碘酮抗心律失常等藥物治療。出院時患者胃脹及肢體無力等癥狀略有緩解,神經系統查體較前無明顯變化。
家系調查:一個家系4代共5例患者(男4例,女1例),分別為先證者(Ⅳ5)及其大伯(Ⅲ1)、父親(Ⅲ3)、表叔(Ⅲ8)及二哥(Ⅳ3),除先證者外均已故。患者家系圖見圖3。
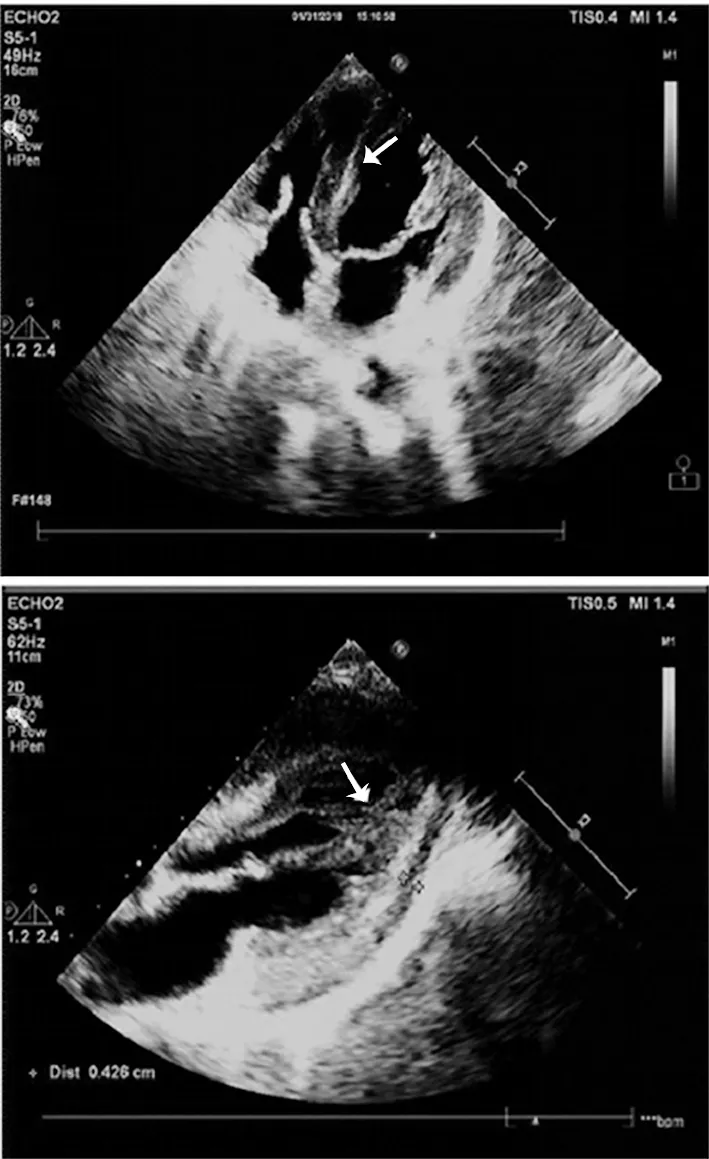
圖1 心臟超聲心動圖示心尖四腔心切面及胸骨旁長軸切面心肌淀粉樣變性(箭頭顯示室壁增厚,回聲減低,)
2 討 論
TTR型FAP是一種罕見疾病,最早于1952年由葡萄牙北部的Andrade報道。據統計此病在葡萄牙及瑞典、日本、巴西相對高發,其發病率在1/10 000 ~1/1 000,其中95%~99%為TTR Val30Met突變,這種突變占家族性淀粉樣多發神經病世界移植登記處(FAPWTR)報道的TTR突變的85%[5]。歐洲的發病率估計為每年0.003/萬,美國發病率估計為1/10萬。
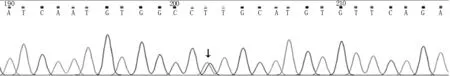
圖2 先證者基因檢測結果:TTR基因測序圖發現c.148G>T(p.V50L)雜合變異(如箭頭所示)
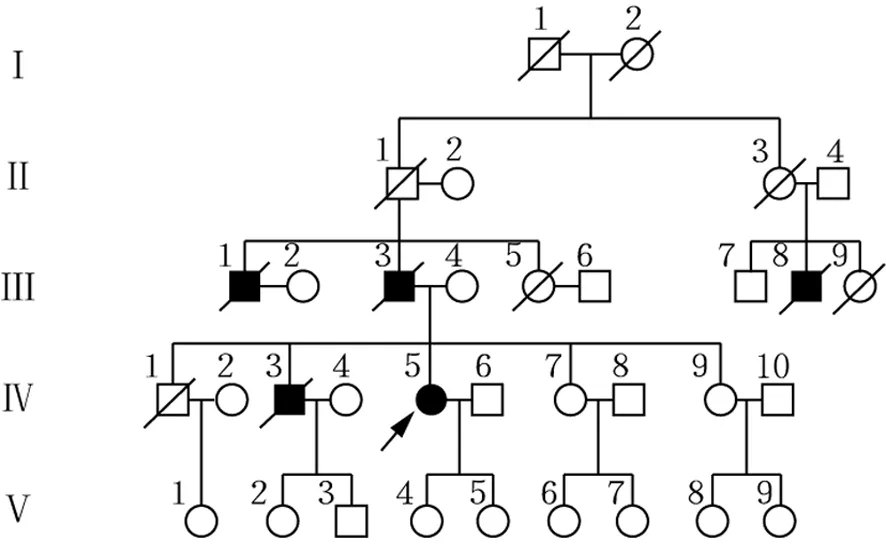
注:□正常男性;○正常女性;/已逝者;■男性患者;●女性患者;→先證者
圖3 FAP患者家系圖譜
而我國東北、福建、香港、臺灣及澳門等地亦有FAP家系報道,其中李延峰等[6]在2001年首次從臨床、病理等方面報道了我國FAP一家系病例。劉晶瑤等[7]在2012年詳細闡述了此病臨床表現及基因表型。由TTR基因c.148G>T突變所致FAP病例十分少見,僅Suhr等[8]曾報道過1例66歲芬蘭病例。而楊碩等[1]在2017年也報道了一家系以足部疼痛為首發癥狀的早發型FAP,填補了我國對TTR基因c.148G>T突變致FAP研究的空缺。
FAP可根據地理分布、致病蛋白及臨床表現的不同分為5型(見表1)。FAPⅠ型(日本、葡萄牙型)是世界最常見的FAP類型,1978年Costa最早證實葡萄牙FAP患者的淀粉樣沉積物為TTR基因點突變所致,其主要臨床特征是緩慢進展的感覺運動性神經病和自主神經功能障礙。通常是從下肢末端(腳趾不適)的感覺神經開始出現異常如皮膚感覺異常,溫覺和痛覺異常較早出現[9]。疾病的中期,感覺缺失呈手套—襪套樣分布并累及運動神經出現肌無力及肌萎縮。自主神經功能障礙癥狀突出,主要損害心臟循環系統、泌尿生殖系統和胃腸道系統,導致心臟傳導衰竭、直立性低血壓、陽痿、惡病質、發作性惡心嘔吐、胃輕癱、腹瀉、便秘或二者交替、排尿困難、性功能障礙及泌汗異常等。后期患者臥床不起或依靠輪椅生存,表現為惡病質,平均存活10年左右[10]。中樞神經損害表現為精神行為異常,腦神經損害及錐體束征。眼睛受累表現包括玻璃體混濁伴進行性視力喪失、小梁阻塞所致慢性開角型青光眼和扇形瞳孔[11]。也可累及腎臟發生漸進性腎衰竭。據報道,超過2/3的TTR基因突變者伴有心肌病[12],典型超聲心動圖表現為心室壁增厚伴顆粒狀或斑點狀心肌,左心室體積小,舒張受限[13]。在心臟淀粉樣變性的突變位點中,Val122Ile在非洲裔美國人群中發病率顯著增高,其中3.0%~3.9% 的突變基因為雜合子[14]。本例家系臨床表現和FAP-I型特別類似,患者的大伯、父親、表叔、二哥都是以消化道癥狀為首發,逐漸出現感覺運動功能癥狀,同時伴有自主神經功能障礙,最后因過于消瘦死于惡病質。而患者除以上癥狀外,出現心功能不全癥狀,同時心電圖及動態心電圖有病理改變,心臟超聲顯示心肌淀粉樣變性。神經肌電圖呈神經元性損害。基因分析發現c.148G>T(p.V50L)雜合變異,可確定診斷FAPⅠ型。
FAP的早期診斷是至關重要的,但由于表型異質性、癥狀出現較晚,診斷延遲和誤診仍很常見。在意大利最近的一項研究中,此病誤診率高達32%,從最初的癥狀到確診約3年,約20%誤診為慢性炎性脫髓鞘多神經病,其次是腰骶神經根病、腰椎管狹窄、副蛋白血癥性神經病和系統性輕鏈淀粉樣變性(AL淀粉樣變性)[15]。對于有FAP家族史的患者,如果接受了基因檢測,組織活檢并不是必須的[11]。本例家系先證者以消化道癥狀為首發表現,后逐漸出現多發性感覺運動神經病,心臟超聲檢查發現彌漫性心肌損害,提示淀粉樣物質沉積,結合其多發性感覺運動神經病、明確家族史,考慮TTR-FAP可能,遂僅行TTR基因檢測,結果提示c.148G>T(p.V50L)雜合變異而得以明確診斷。
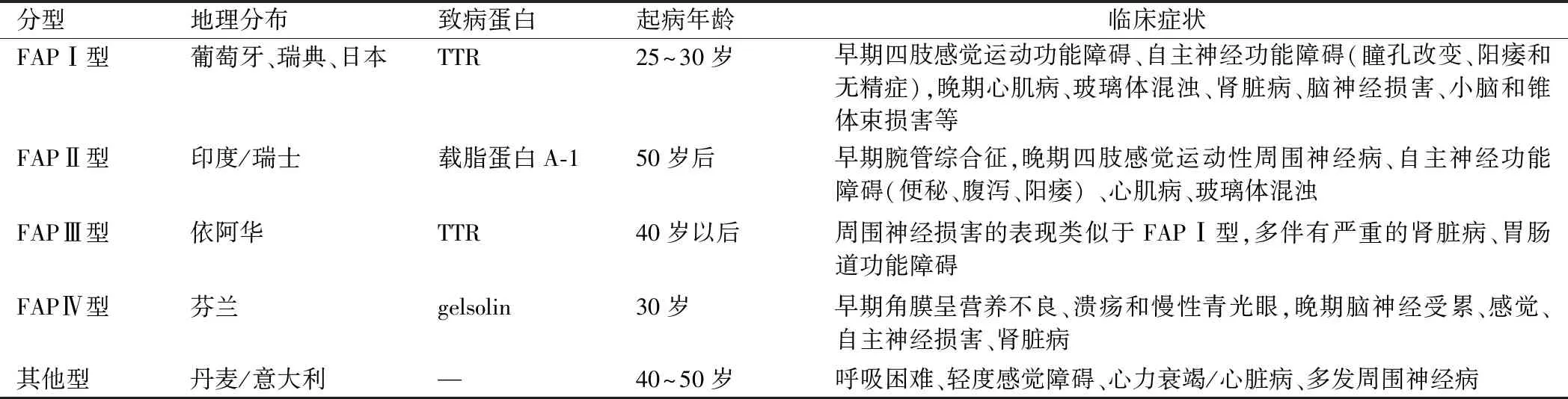
表1 家族性淀粉樣周圍神經病分型
但因先證者曾祖、祖父一代去世早,時間久遠,家人無法回憶當時情況,不能確定此2代人患病情況,但此家系自第Ⅲ代開始表現為明確的常染色體顯性遺傳,符合TTR-FAP臨床特征。
TTR是一種同源四聚體蛋白,主要產生于肝臟,少量產生于脈絡膜叢和視網膜細胞[16],因此,對于早發患者可行肝移植(LT)治療,LT是TTR-FAP的第一個治療方案,其15年生存率接近80%[17-18],而未行肝移植的患者10年生存率僅為 56.1%[19]。目前一種轉甲狀腺素蛋白穩定劑藥物(氯苯唑酸),通過與甲狀腺素結合位點結合穩定突變的TTR四聚體,防止其分解成單體,減少淀粉樣蛋白形成,可能改善神經系統和心臟情況的進展[20-21]。二氟尼柳是20世紀70年代開發的非甾體抗炎藥(NSAID),是體外形成TTR淀粉樣蛋白纖維的強抑制劑,它可以防止突變TTR四聚體的分離、錯折疊和錯組裝。Berk等[22]的一項多中心研究表明,二氟尼柳可抑制多神經病進展,提高TTR-FAP患者不同疾病階段的生活質量。目前的一種基因沉默療法[使用小干擾RNA (siRNA)或反義寡核苷酸ASO],通過強烈降低突變體和野生型TTR血漿水平,可抑制TTR蛋白的產生[23]。目前正在研究淀粉纖維的靶向藥物抗血清淀粉樣蛋白P(SAP)制劑或抗TTR抗體,其目標在于清除組織中已經存在的淀粉樣蛋白沉積,以減少淀粉樣蛋白負荷,最終恢復器官功能[24]。采用脂質納米粒子技術的ALN-TTR01和ALN-TTR02是一種系統性RNA干擾療法,旨在抑制TTR蛋白的表達,防止淀粉樣蛋白的形成[25]。對于所有TTR-FAP突變攜帶者,無論有無癥狀,都應定期進行神經、心臟、腎臟和眼部檢查。神經檢查包括感覺、運動和反射評估,肌電圖、體位血壓和心率變異性檢測。心臟篩查包括進行心電圖和超聲心動圖,測量BNP或氨基末端腦鈉肽前體(NT-pro-BNP)。除此之外,對癥治療也很重要,針對神經痛可以應用卡馬西平、加巴噴丁、普瑞巴林等藥物;針對心臟受累后出現傳導阻滯可以安裝起搏器;針對體位性低血壓等癥狀可以加用藥物及調整生活方式控制改善癥狀[26]。
綜上,TTR-FAP是一種罕見的、TTR基因突變所致的、表現為多發性感覺運動神經病的全身性疾病,部分患者可能以消化道癥狀等自主神經損害為首發癥狀。對于具有家族史的、以多發性感覺運動神經病為主要表現的患者,如超聲提示心臟淀粉樣物質沉積,應考慮TTR-FAP診斷可能,行TTR基因檢測可明確。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
