大鼠Pdcd4基因真核載體的構建與表達
2019-12-02 01:13:48呂社民劉海燕金巧艷
醫學研究雜志 2019年10期
鐘 波 李 靜 呂社民 侯 偉 劉海燕 金巧艷 魏 偉
程序性細胞死亡因子4(programmed cell death 4,Pdcd4)基因最初被發現與細胞的程序性死亡有關[1]。該基因在大鼠定位于1q55, cDNA全長為1410bp,所編碼的蛋白質共含個469氨基酸。筆者在之前的研究中應用抑制性消減雜交技術篩選出Pdcd4作為抗原誘導肺部炎癥上調表達的差異表達基因[2]。為進一步研究該基因的功能,以E3大鼠為研究對象,克隆其肺組織Pdcd4基因并構建pEGFP-C1-Pdcd4重組真核載體,為揭示該基因的功能建立基礎。
材料與方法
1.菌株與載體:所用感受態DH5α大腸桿菌菌種、真核表達載體pEGFP-C1均為西安交通大學醫學部基礎醫學院分子生物學系實驗室保存。本研究選用含有綠色熒光蛋白及Hind Ⅲ與Xho Ⅰ酶切位點的pEGFP-C1質粒作為重組載體。其結構與多克隆位點如圖1所示。
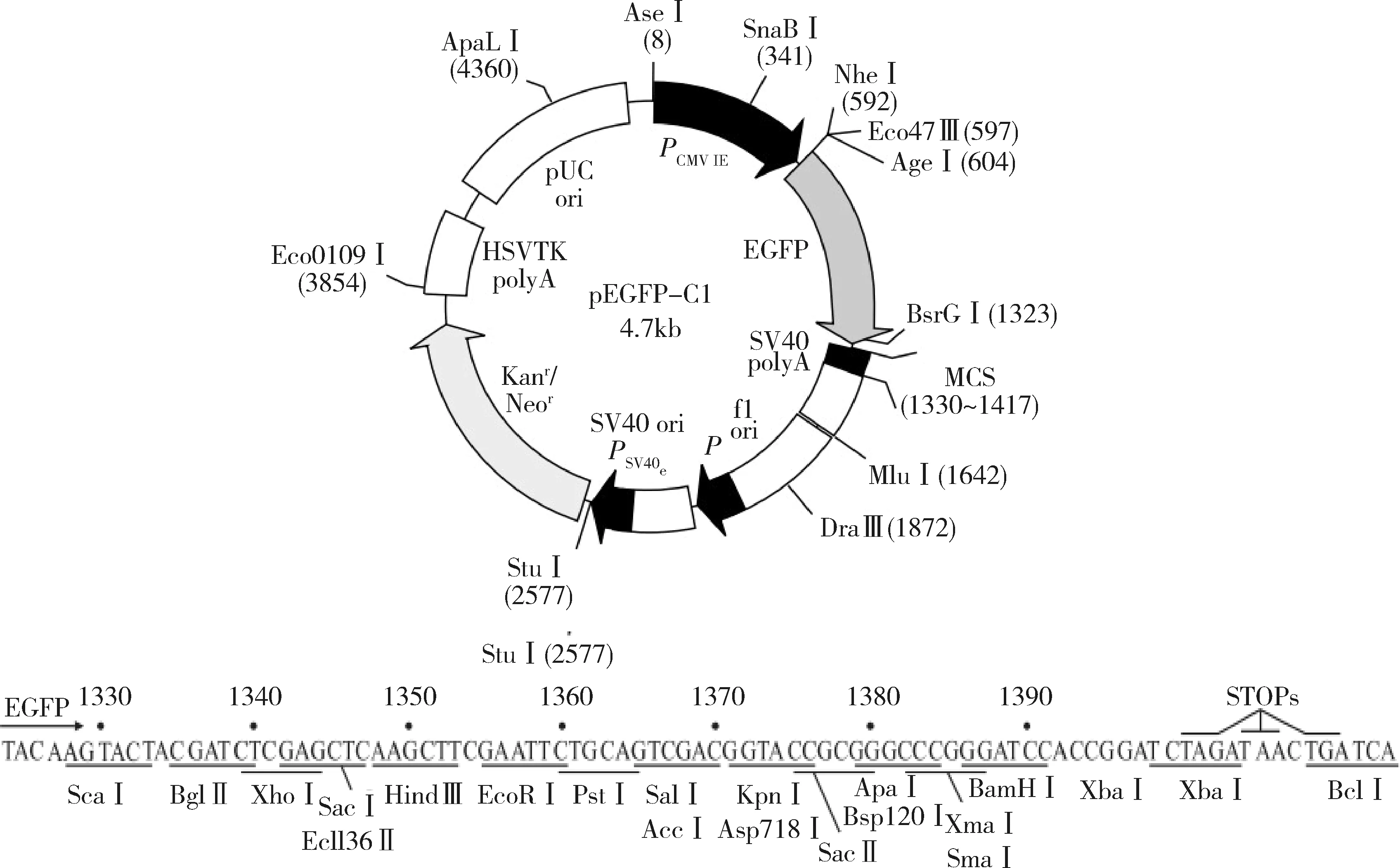
圖1 pEGFP-C1載體結構和多克隆位點示意圖
2.細胞與實驗動物:本研究使用大鼠巨噬細胞系NR8383細胞與8周齡近交系E3大鼠(SPF級動物房繁育),均為西安交通大學醫學部基礎醫學院生物化學與分子生物學系所保存。
3.試劑:Trizol?Reagent、Lipofectamine 2000購自美國 Invitrogen公司,RevertAidTM First Strand cDNA Synthesis Kit、限制性內切酶Hind Ⅲ與Xho Ⅰ購自加拿大Fermentas公司,質粒提取試劑盒購自北京天根生物技術公司,膠回收試劑盒購自北京博大泰克生物基因技術有限責任公司,T4 DNA連接酶購自美國Sigma公司,胰蛋白胨及酵母提取物購自英國Oxoid公司,高保真Taq酶、DNA Marker購自日本 TaKaRa 公司,F12K培養基、胎牛血清購自美國HyClone公司,其他試劑均為國產分析純。6孔板購自美國Corning公司,倒置相差顯微鏡及正置熒光顯微鏡購自日本Olympus公司。
4.引物:PCR引物均由北京三博遠志生物技術有限責任公司合成,序列信息如表1所示。

表1 PCR引物信息
5.實驗方法
(1) E3大鼠肺組織總RNA提取:將大鼠稱重,按1g/kg體重烏拉坦(質量體積比為20%)的劑量麻醉大鼠;經腹主動脈取血,從甲狀軟骨處剪斷氣管,分離肺組織,放入研缽中加液氮徹底研磨,加Trizol室溫放置5min,加入1/5體積三氯甲烷,混勻15s,室溫靜置10min,4℃ 12000r/min離心20min,取上層水相,加入等量預冷的異丙醇,混勻后-20℃放置2h,4℃ 12000r/min離心15min。棄上清,加入1ml預冷的75%乙醇,重新懸浮沉淀,7500r/min離心5min。棄上清,使剩余乙醇徹底揮發。20μl DEPC-H2O溶解RNA,所得RNA溶液保存在-80℃低溫冰箱備用。微量核酸/蛋白定量儀上測定總RNA濃度,RNA電泳鑒定總RNA的完整性。
(2) cDNA反轉錄與PCR擴增Pdcd4 cDNA全長:按照RevertAidTM First Strand cDNA Synthesis Kit說明書操作,反應體系oligo(dT)18引物1μl,RNA 2μg,DEPC處理水加至總體積12μl,混勻、離心。70℃孵育5min后于冰上放置,加入5×反應緩沖液4μl,dNTP(10mmol/L)2μl,M-MLV 反轉氯酶1μl, 核糖核酸酶抑制劑1μl。混勻、離心,42℃孵育1h,70℃ 10min終止反應。建立Pdcd4全長擴增的反應體系:cDNA 0.5μl,Pdcd4克隆上游引物1μl,Pdcd4克隆下游引物1μl,高保真Taq酶0.5μl,5×Taq酶緩沖液10μl,dNTP 4μl,H2O 33μl。反應條件95℃10min,30個循環95℃ 45s、57℃ 45s、72℃ 1.5min,循環結束,72℃ 延長10min。生成的PCR擴增產物用于后續酶切實驗。
(3)重組質粒的構建與鑒定:①建立酶切PCR擴增產物反應體系:Hind Ⅲ 2μl,Xho Ⅰ 2μl,10×M緩沖液 3μl,PCR擴增產物20μl,H2O 3μl。建立pEGFP-C1質粒載體反應體系:Hind Ⅲ 2μl, Xho Ⅰ 2μl,10×M緩沖液 5μl, pEGFP-C1質粒 30μl, H2O 11μl。37℃酶切過夜。將雙酶切好的目的DNA片段和載體電泳鑒定,之后用膠回收試劑盒回收;②建立連接反應體系:DNA片段8μl,pEGFP-C1載體4μl,T4 DNA連接酶1μl,10×連接緩沖液 2μl,H2O 5μl,16℃水浴連接過夜。取5μl連接產物加入200μl DH5α感受態細胞中,冰上放置30min。42℃熱激90s,冰上放置5min。加入LB培養基1ml,37℃搖床150r/min復蘇1h。離心1min后,棄掉上清900μl,剩下的菌液混勻后涂布在含卡那霉素的LB固體培養基平板上,菌液吸收后,倒置放入37℃培養箱中過夜培養。挑選單克隆菌落在LB培養液(含卡那霉素)中37℃過夜擴增。次日經菌液 PCR 檢測鑒定插入片段,并按照質粒提取試劑盒的說明步驟提取質粒。將經菌液 PCR 檢測鑒定有插入片段的質粒測序送交生物公司測序,后至 NCBI數據庫行BLAST比對,確定插入片段無突變,表明克隆構建成功。
(4)細胞轉染:轉染共分3組:空白對照組、pEGFP-C1組和pEGFP-C1-Pdcd4組,每組3個復孔。6孔板中每孔接種8×105個NR8383細胞,加2ml 10%胎牛血清F12K培養基培養,接種24h后將培養基換為2ml不含胎牛血清的F12K培養基。取無菌EP管加入脂質體5μl和245μl無血清培養基,混勻后37℃孵育5min。取無菌EP管,加入2μg質粒DNA,加入無血清培養基至總體積為250μl,每種質粒配3份,空白對照組只加無血清培養基250μl,混勻后37℃孵育5min。各取一管脂質體與一管質粒混合,使總體積為500μl,瞬時離心,37℃孵育20min。將脂質體和質粒的混合物加入6孔板對應的孔中。24h后倒置熒光顯微鏡下觀察綠色熒光的表達以確定轉染效率。同樣的轉染共重復3次,將3次的結果取平均值為最終結果。
(5)總RNA提取、cDNA反轉錄與RT-qPCR:轉染24h后倒置熒光顯微鏡下可以觀察到明顯的綠色熒光表達,收集細胞于1.5ml EP管中,1000r/min離心10min,離心后取一半沉淀加入1ml Trizol并吹打數次。按前述步驟提取總RNA,-80℃保存備用。測定總RNA濃度,電泳鑒定總RNA的完整性。按照前述的方法反轉錄合成cDNA,產物保存于-20℃備用。在mRNA水平檢測細胞中Pdcd4在體RNA干擾的效率,反應體系:20倍稀釋的cDNA 4μl、SYBR?Premix Ex TaqTM Ⅱ 4μl、Pdcd4上游引物0.5μl、 Pdcd4下游引物0.5μl、H2O 1μl,混勻、瞬時離心。反應條件95℃ 1min,40個循環95℃ 10s,退火溫度30s,72℃ 30s,熔解曲線41個循環,55~95℃ 30s。引物信息見表1,選GAPDH為內參,結果采用ΔΔCt法計算相對基因表達量。
(6)Western blot法檢測Pdcd4的表達水平:細胞離心后取另一半沉淀加入含有蛋白酶抑制劑的RIPA細胞裂解液,冰上放置30min,12000×g4℃離心20min,取上清,BCA法測定蛋白濃度。常規方法制備分離膠、濃縮膠。將蛋白樣品與5×上樣緩沖液混合,100℃保溫5min使蛋白變性,取50μg蛋白上樣,電泳分離后將蛋白質從凝膠中轉移到NC膜上。5%脫脂奶粉室溫封閉1h。Pdcd4(山羊抗大鼠)一抗使用濃度為1∶200,β-actin使用濃度為1∶500,4℃孵育過夜。1×TBST洗膜3次×15min。Pdcd4(兔抗山羊)二抗使用濃度為1∶5000,β-actin使用濃度為1∶1000,26℃孵育1h。1×TBST洗膜3次×15min。ECL顯色,發光。
結 果
1.肺組織總RNA電泳鑒定:提取的肺組織總RNA經微量核酸蛋白定量儀和1%瓊脂糖凝膠電泳鑒定。總RNA電泳結果顯示總RNA條帶清晰,28S∶18S約為(1.5~2.0)∶1.0(圖2)。
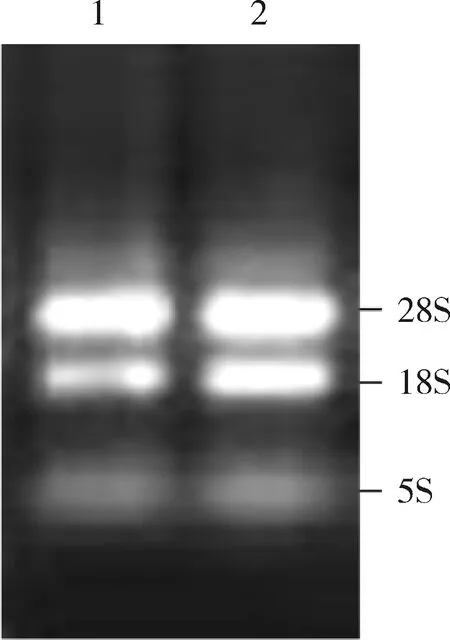
圖2 電泳檢測肺組織總RNA1.2μl大鼠肺組織總RNA上樣;2.3μl大鼠肺組織總RNA上樣
2.重組質粒的構建與鑒定:首先以大鼠肺組織cDNA為模板,用特異性引物PCR獲得Pdcd4的cDNA全長,與pEGFP-C1質粒載體同時雙酶切,雙酶切后的Pdcd4 cDNA片段大小約1400bp,pEGFP-C1載體在酶切之前表現為兩種條帶,酶切之后形成線性化條帶,大小均一(圖3A)。
將pEGFP-C1載體片段和Pdcd4 cDNA連接后轉化入大腸桿菌,涂平板過夜培養后見平板長滿菌落,隨機挑取10個單獨菌落行過夜培養擴增,次日行菌液 PCR 檢測,部分克隆結果見陽性條帶(圖3B),片段大小約1400bp,這些克隆為陽性克隆,取相應質粒送公司測序,測序結果顯示未見變異重組質粒構建成功。

圖3 雙酶切與菌液PCR電泳結果A.Pdcd4 全長cDNA與pEGFP-C1質粒載體的雙酶切電泳鑒定:1.Pdcd4 全長cDNA雙酶切前; 2.Pdcd4 全長cDNA雙酶切后; 3.pEGFP-C1質粒載體雙酶切前;4.pEGFP-C1質粒載體雙酶切后;M.DL2000 DNA marker;B.菌液 PCR產物電泳鑒定:1、2.隨機排選不同克隆的菌液PCR產物;M.DL2000 DNA marker
3.NR8383細胞轉染:將構建好的重組pEGFP-C1-Pdcd4質粒和對照空載體質粒pEGFP-C1分別轉染大鼠巨噬細胞系NR8383細胞, 24h后于熒光倒置顯微鏡下觀察轉染效率,可以看到細胞中有明顯的綠色熒光的表達(圖4),轉染成功。

圖4 細胞轉染后綠色熒光蛋白的表達(×100)A、C.細胞在光鏡下的形態;B、D.細胞在熒光顯微鏡下的形態及綠色熒光蛋白的表達
4.檢測Pdcd4的表達:轉染24h后顯微鏡下可見細胞內有明顯綠色熒光蛋白的表達,收集細胞,通過real-time PCR和Western blot法分別檢測轉染后細胞Pdcd4在mRNA水平與蛋白質水平的表達變化,在轉染pEGFP-C1-Pdcd4質粒后,NR8383細胞Pdcd4的表達與對照空載體pEGFP-C1組比較明顯上調(圖5)。
討 論
Pdcd4基因是1995年由Chen等[3]在小鼠體內發現的與細胞凋亡有關的基因,在細胞發生程序性死亡時被誘導產生。Pdcd4在不同的種屬中有不同的名稱,如在小鼠又名MA-3蛋白、拓撲異構酶抑制子抑制蛋白,在人類又被稱為腫瘤性轉化抑制劑、核抗原H731、197/15a蛋白,在大鼠被稱為死亡上調基因蛋白[4]。

圖5 轉染后NR8383細胞Pdcd4的表達A.real-time PCR;B.Western blot法
Pdcd4蛋白質最顯著的結構特征是在其C端有兩個重要的螺旋結構域MA-3,Pdcd4 MA-3功能域通過一個保守表面區與真核翻譯起始子eIF4A的N端功能域相互作用、發生結合,從而抑制核糖體復合物的形成和蛋白質的合成,促進細胞凋亡[5]。Waters等[6]對Pdcd4的三維空間結構進行了研究,解析了Pdcd4的C端MA-3結構域的結構,找到了它與eIF4A相互作用的特征,并且對比了來自單個功能域和串聯的MA-3部位的核磁共振譜的特點,發現Pdcd4 MA-3(C)由三螺旋-轉角-螺旋發卡結構組成。
本實驗克隆了E3大鼠肺組織Pdcd4基因,將其CDS序列克隆至pEGFP-C1質粒中構建了重組真核載體pEGFP-C1-Pdcd4,通過酶切鑒定、基因測序和序列比對,驗證了目的基因的正確性。pEGFP-C1-Pdcd4成功轉染大鼠巨噬細胞系NR8383細胞并表達出綠色熒光蛋白,增加NR8383細胞中Pdcd4基因的表達。
有多種miRNA可以影響Pdcd4的表達,主要包括miR-21、miR-141、miR-16、miR-96、miR-503等[7~11]。這些miRNA通過對Pdcd4表達的調控,最終影響腫瘤細胞的增殖、浸潤、轉移及對化療藥物的反應等。近年來發現長鏈非編碼RNA(long non-coding RNAs,LncRNAs)也通過對Pdcd4表達的干預參與影響腫瘤的進展。例如,LncRNA CASC15可以在Pdcd4基因的啟動子區募集zeste基因增強子同源物、增加組蛋白 H3K27的水平,沉默Pdcd4的表達,進而促進黑色素瘤的進展[12]。
綜上所述,目前關于Pdcd4基因功能的研究主要集中于以上這些促進凋亡、抑制腫瘤的方面,筆者之前的研究發現Pdcd4是抗原誘導肺部炎癥上調表達的差異表達基因,其差異表達來源于肺泡的巨噬細胞[2]。對于該基因在炎癥中的作用文獻報道較少,關于其在巨噬細胞中作用的文獻更少。本研究成功克隆獲得E3大鼠Pdcd4基因編碼序列,即可直接用于后期Pdcd4基因的功能探索及上調表達等研究。
