基于第一性原理的層狀ZnO結構和化學鍵研究
2019-12-13 08:24:09蘇亞拉
現代商貿工業 2019年34期
蘇亞拉
摘 要:第一性原理系統的研究了層狀ZnO的結構和化學鍵的特性。計算的結果表明層狀ZnO既存在共價鍵又有離子鍵特性與其他人的結論相一致。
關鍵詞:層狀ZnO;幾何結構;化學鍵
中圖分類號:TB 文獻標識碼:A doi:10.19311/j.cnki.1672-3198.2019.34.089
0 引言
早在2010年,Pueyo等人就利用Zn4O4為前驅體成功地合成了層狀六角結構的ZnO相。室溫下,它的帶隙能為~3.5eV,比纖鋅礦ZnO略大。通過結構分析表明,這種層狀ZnO的a-b面上Zn-O鍵長1.79,小于纖鋅礦(B4)ZnO中Zn-O鍵長1.94。這種層狀ZnO表現出與纖鋅礦ZnO相近的光學帶隙,有可能成為一種新型的光電子材料。Rakshit和Mahadevan首次通過第一性原理計算討論了這種層狀ZnO的穩定性。Molepo and Joubert預測這種層狀ZnO可以通過纖鋅礦ZnO高壓相變獲得,相變壓力24.6GPa,但尚需進一步的實驗驗證。2015年,Wang等人利用超軟贗勢計算了這種層狀ZnO的電子結構和光學性質。然而,對于層狀ZnO物理特性的研究仍然沒有深入的探討,尤其是它的化學鍵。本文基于第一性原理密度泛函理論研究層狀ZnO的化學鍵屬性。
1 計算方法
基于密度泛函理論,計算采用模守恒贗勢交換關聯相互作用采用廣義梯度近似(GGA)泛函。Zn和O的電子構型分別為3d104s2和2s22p4。能量優化采用 BFGS算法,平面波的截斷能為900eV。B4相和層狀Bk相ZnO的全布里淵區k點求和為7×7×4。測試結果表明進一步增加平面波的截斷能和k點數目幾乎對計算結果沒有影響。
2 結果與討論
2.1 結構與參數
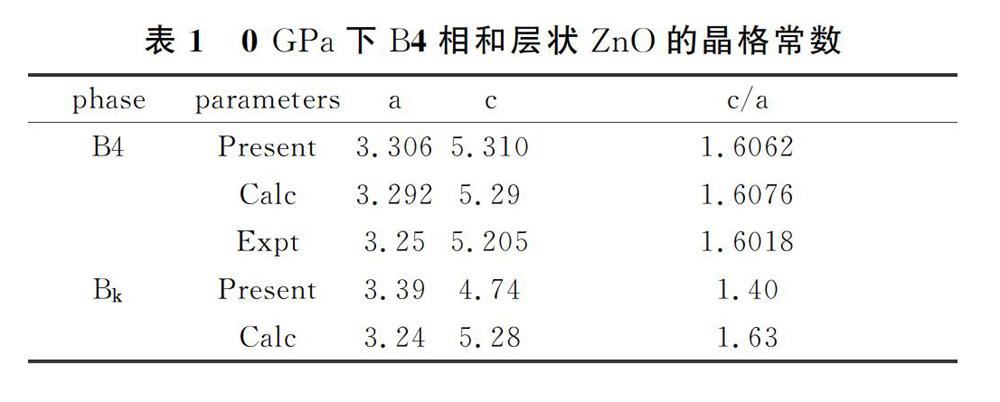
為了評價計算結果的可靠性,我們首先幾何優化了0GPa下B4相和層狀ZnO的晶格常數(a,c,c/a)并與實驗結果和他人的理論結果進行了比較,如表1所示。從表中可以看出,B4相和層狀ZnO的計算結果與實驗結果和他人的計算結果基本一致,基本參數a、c、c/a的計算誤差小于2%,說明我們采用的計算方法和參數設置是可靠的。
2.2 分析化學鍵
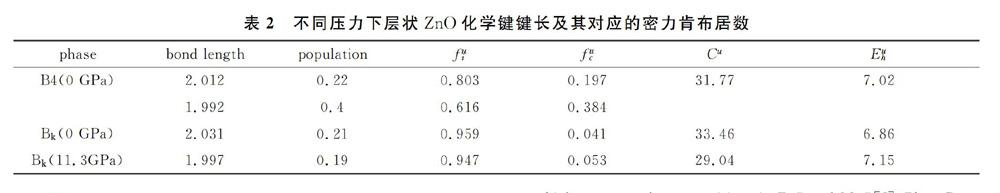
表1列出了0 GPa下B4相以及0 GPa和11.3GPa下層狀ZnO化學鍵鍵長及其對應的密力肯布居數。0 GPa下B4相計算的鍵長與前人的結果基本相同。從表中可以看出,0 GPa下B4相的鍵長比0 GPa下層狀ZnO化學鍵要長對應的密力肯布居數基本上沒有變化。然而,在相轉變壓強下,層狀ZnO化學鍵的鍵長減小。為了判斷是否是離子鍵還是共價鍵,這可通過化學鍵理論分析。張等人提出過晶體的化學鍵理論,它是以化學鍵鍵能為基礎的半經驗方法。計算背景為:首先通過結構分析,將復雜的分子化學式分解成簡單的二元分子式的組合,然后可以計算離子鍵和共價鍵的代表參數。對于ZnO本身就是最簡單的二元分子式,如表2所示,其中代表鍵長。從表中數據可以看出,0 GPa下B4相計算得到化學鍵的異極化能大于同極化,這表明它是以離子性為主,與Phillips結論一致。但是我們計算的離子性標定為0.803大于Phillips的離子性標定0.616,這歸因于計算的平均磁化率不同有關。對于0 GPa下層狀ZnO,同樣是以離子性為主。相轉變后離子性標定減小,說明隨著壓強的增大離子性降低,這與我們以前的結論相同。通過計算得到不同化學鍵的離子性結果也能證明這一結論。總之,層狀ZnO與B4相的ZnO一樣,即表現出離子鍵又表現共價鍵特性。
3 結果
利用密度泛函理論研究了層狀ZnO的幾何結構參數和化學鍵的特性。根據化學鍵理論計算結果表明層狀ZnO既存在共價鍵又有離子鍵特性。
參考文獻
[1]Pueyo C L,Siroky S,Landsmann S,et al.Molecular precursor route to a metastable form of zinc oxide[J].Chem.Mater,2010,(22):4263.
[2]Rakshit B and Mahadevan P.Stability of the bulk phase of layered ZnO[J].Phys.Rev.Lett,2011,(107):085508.
[3]Molepo M P and Joubert D P.Computational study of the structural phases of ZnO[J].Phys .Rev.B,2011,(84):094110.
[4]Wang Q B,Zhou C,Wu J,et al.GGA+U study of the electronic and optical properties of hexagonal BN phase ZnO under pressure[J].Comp.Mater.Sci,2015,(102):196.
[5]Hamann D R,Schluter M,and Chiang C.Norm conserving pseudopotentials[J].J.Phys.Rev.Lett,1979,(43):1494.
[6]Perdew J P,Burke K,and Ernzerhof M.Generalized gradient approximation made simple[J].Phys.Rev.Lett,1996,(77):3865.
[7]Jaffe J E,Snyder J A,and Lin Z.LDA and GGA calculations for high-pressure phase transitions in ZnO and MgO[J].Phys.Rev.B,2000,(62):1660.
[8]Desgreniers S.Structural and compressive parameters high density phases of ZnO[J].Phys.Rev.B,1998,(58):14102-14105.
[9]Sun J and Wang H T.Ab initio investigations of optical properties of the high-pressure phases of ZnO[J].Phys Rev B,2005,(71):125132.
[10]Fan C,Wang Q,Li L X,et al. Bulk moduli of wurtzite,zinc-blende,and rocksalt phases of ZnO from chemical bond method and density functional theory[J].Appl.Phys.Lett,2008,(92):101917.
[11]Zhang S,Li H,Li L,et al. Calculation of bulk modulus on carbon nitrides with chemical bond method[J].Appl Phys Lett,2007,(91):251905.
[12]Phillips J C.Bonds and Bands in semiconductors Academic[Z].New York,1973.
[13]Vechten J A V.Quantum Dielectric Theory of Electronegativity in Covalent Systems.Electronic Dielectric Constant[J].Phys Rev,1969,(182):891.
[14]Serrano J,Romero A H,Manjon F J,et al. Pressure dependence of the lattice dynamics of ZnO:An ab initio approach[J].Phys.Rev.B,2004,(69):094306.
