SDF-1/CXCR4及下游信號通路在骨性關節炎病程中的作用
2019-12-25 09:04:12何映紅李彥林向耀宇陳泳佳楊驍
實用醫學雜志 2019年22期
關鍵詞:信號
何映紅 李彥林 向耀宇 陳泳佳 楊驍
昆明醫科大學第一附屬醫院運動醫學科(昆明655031)
近年來,骨性關節炎(osteoarthritis,OA)在老年人群體中的發病率逐年上升,且逐漸年輕化,傳統治療方法只能通過抑制炎癥因子、改善關節腔微環境、置換關節等方式從癥狀及表觀上間接治療,但效果有限,未能從根本上阻斷OA 發展。隨著對骨性關節炎和類風濕性關節炎(rheumatoid arthritis,RA)的不斷深入研究,發現基質細胞衍生因子-1/CXC 趨化因子受體4 型(stromal cell derived factor-1/ chemokine receptor-4,SDF-1/ CXCR4)信號通路在這兩種疾病進程中發揮重要作用。OA 通常被稱為退行性關節病,但這種說法并不完整,因為OA 不僅僅是一個磨損過程,而是由受累關節內的大量炎癥介質驅動的關節組織異常重塑,致使關節軟骨退化,同時軟骨細胞不能修復關節軟骨基質,導致組成和結構的變化。然而,關節軟骨不是該疾病中唯一受影響的關節內組織。OA 軟骨下骨通過間充質干細胞的增殖和軟骨細胞的分化表現出重塑和微結構變化,使軟骨下骨增厚、關節邊緣中骨贅形成[1]。此外,免疫細胞滲入OA 滑膜,增強促炎介質的產生,這種滑膜炎也會導致軟骨細胞基質失調,從而使軟骨退化,半月板損傷及韌帶退化[2],導致疼痛、畸形和功能喪失。而所有的這些過程都涉及有關信號分子的傳遞。目前的研究[3-4]表明,SDF-1/CXCR4 在骨性關節炎的進展中發揮重要作用,SDF-1 與其受體結合后使其C 末端的酪氨酸殘基被磷酸化,激活多種下游信號通路,從而促進OA 的發生與發展。
1 MAPK 在OA 中的作用
磷酸特異性免疫印跡證實SDF-1α 激活軟骨祖細胞中的所有三種絲裂原活化蛋白激酶通路(mitogen activated protein kinase pathway,MAPK),包括ERK1/2 通路,JNK 通路和P38 通路[5]。創傷后關節炎的研究發現SDF-1 可激活p38 和ERK1/2通路,它們是激活蛋白-1(activator protein-1,AP-1)活性的有效誘導劑,AP-1 對酒石酸抗性酸性磷酸酶(tartrate-resistant acid phosphatas,TRAP),基質金屬蛋白酶(matrixmetalloproteinase-9,MMPs)和組織蛋白酶K(cathepsin K,CK)的表達至關重要[6],而TRAP 和CK 是破骨細胞的特異性標志物,也就是說,SDF-1 可以通過激活AP-1 增加破骨細胞的分化,促進骨質吸收,降低骨質鈣含量,從而增加骨質疏松的風險[3]。在亞細胞水平,SDF-1 激活p38和Erk1/2 信號通路,其一方面誘導細胞周期蛋白D1 促進軟骨細胞增殖,另一方面激活Runx2,增加MMP-13 生成的量,促進軟骨細胞向末端成熟和凋亡[7]。顳下頜關節OA的研究證明激活ERK 信號通路有助于誘導基質金屬蛋白酶的產生[8]。Erk1/2信號通路的激活可以誘導許多蛋白激酶級聯反應,并將細胞外信號傳遞到細胞中[9]。磷酸化的ERK1/2 進入細胞核并觸發轉錄因子和MMPs(包括MMP-3 和MMP-13)的活性,這有助于OA 病程啟動和進展過程中細胞外基質的降解,引起一系列可調節細胞凋亡的細胞反應[10]。此外,p38 的激活,刺激軟骨細胞分泌基質金屬蛋白酶13(MMP-13),MMP-13 可降解人OA 中的Ⅱ型膠原蛋白和聚集蛋白聚糖,從而誘導軟骨細胞死亡[11]。SDF-1 激活p38 通路使得血管內皮生長因子(vascular endothelial growth factor,VEGF)表達上調,促進滑膜血管形成,增加滑膜炎癥因子和巨噬細胞浸潤,加重OA 的炎癥[5]。同時軟骨及軟骨下骨血管形成增加,加劇軟骨細胞肥大及軟骨下骨贅形成增加,加重OA 晚期關節退變的速度[12]。但軟骨祖細胞的條件培養試驗表明VEGF 表達基本上僅被p38 特異性抑制劑阻斷,而不能被ERK 和JNK 特異性抑制劑阻斷,所以軟骨祖細胞中的VEGF 表達反應依賴于p38 活化,但不依賴于ERK 或JNK 通路激活[5]。MAPK 級聯信號通路(ERK1/2 和p38)信號通路激活后,可以調節環加氧酶2的表達[10],COX-2促進花生四烯酸轉化生成前列腺素2,并在關節中誘導關節炎發生[13],故PGE2 被認為是關節炎病癥中炎性疼痛的主要原因[14]。
2 PI3K/AKT 在OA 發展中的作用
CHEN 等[15]研究發現:SDF-1α 激活CXCR4 受體并引起磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/Akt 途徑的激活,PI3K/Akt 在人OA滑膜成纖維細胞的IL-6 表達中起重要作用,該途徑增強c-Jun 與AP-1 位點的結合并導致IL-6 的反式激活,使IL-6表達的上調,IL-6是3種主要的炎性細胞因子之一,和IL-1β和TNF-α一起,與炎癥引起的軟骨損傷有關,IL-6 通過作用于軟骨生理學的合成代謝和分解代謝機制而對軟骨造成損害[16]。PI3K 抑制劑能夠有效抑制SDF-1a 指導的IL-6 產生,但MAPK抑制劑卻無此抑制作用,結果進一步證實了PI3K/Akt 的顯性失活突變體,抑制了SDF-1a對IL-6 產生,而不是通過MAPK 途徑抑制IL-6 表達[15]。此外,IL-6 還被證明是促進破骨細胞分化的趨化因子[17]。
PI3K 還可能調節軟骨細胞的凋亡:在壞死性凋亡過程中通過PI3K 的自磷酸化使混合譜系激酶結構域樣蛋白MLKL 磷酸化導致MLKL 從細胞質轉運至質膜[18],促使質膜碎片化,促進凋亡。然而,也有研究[19]證明PI3K/Akt 信號傳導被認為是NF-κB 信號傳導途徑的主要上游元件,也就是說PI3K/AKT 是通過調節NF-kB 信號通路來增強軟骨細胞凋亡的。細胞凋亡與OA 組織中軟骨破壞程度和基質分解代謝高度相關[20]。與MAPK 級聯信號通路一樣,PI3K/AKT 信號通路激活后,可以調節環加氧酶2 的表達[10],促進PEG2 關節炎關節中誘導,加重關節疼痛[13,21]。同時研究證明,PI3K/AKT 信號通路和MAPK 級聯信號通路(ERK1/2 和p38)激活后,可使活性氧(reactive oxygen species,ROS)生成增加,而諸多證據表明人類各種疾病中的細胞損傷導致氧化應激增加ROS 產生,這些影響是通過與MMPs 的相互作用介導的[22]。在OA中活性氧參與調節涉及軟骨退化的生化因子的產生,它們可能通過激活MMPs減少基質成分合成直接或間接地對所有基質組分造成損害。
3 NF-κB 在OA 發展中的作用
在體外軟骨細胞培養試驗中也發現:SDF-1/CXCR4 軸可能是軟骨細胞中核轉錄因子-κB(Nuclear Factor-κB,NF-κB)信號傳導的其中一個主要上游調節因子[7]。活化B 細胞的NF-κB 家族在廣泛的生物學過程中具有重要作用,包括細胞存活,增殖,分化,凋亡,衰老,炎癥和免疫反應[23]。NFκB 信號通路通過各種作用廣泛參與OA 病理生理學,并在衰老和炎癥期間在OA 軟骨細胞中被激活。NF-κB 途徑是致細胞外基質分解代謝作用的中樞調節劑,介導軟骨細胞的炎癥反應中的關鍵事件并導致細胞外基質損傷和軟骨侵蝕[24]。
NF-κB 途徑對誘導各種炎癥相關因子至關重要:肽聚糖和LPS(TLR-2 和4 的配體),以依賴于NF-κB 的方式分別刺激軟骨細胞增加MMP-1,3 和13 的表達,以及上調NO 產生[25]。而NO 可以激活MMPs,增加蛋白多糖降解,減少IL-1 受體拮抗劑的產生并介導軟骨細胞凋亡[26]。此外,根據劑量和特定的氧化還原衍生物的研究,NO 還可以對OA 進展產生負面和正面影響,特定的NO 衍生物維持NF-κB 活化以響應促炎刺激和其他形式的NO 抑制其誘導[27]。除此之外,NF-κB 通路還可誘導IL-6,IL-1,TNFα,COX-2 等軟骨細胞表達炎癥相關基因[28],這些誘導的細胞因子進一步激活信號級聯反應。COX-2 參與導致PGE2 合成的連續酶促反應,PGE2 通過與關節組織中存在的EP2 和EP4 PGE2 受體結合,在軟骨退化和OA 進展中起重要作用,導致軟骨退化增加和OA 軟骨中的軟骨細胞肥大[29]。PGE2 還參與IL-1β 誘導的MMP-2 表達和活化[30],并誘導關節軟骨細胞凋亡。在這些促炎因子中,IL-1β 和TNF-α 主要參與炎癥,通過引發一系列導致分解代謝增加和軟骨細胞肥大的事件在軟骨降解中發揮重要作用。
在體外軟骨細胞培養研究中還發現:SDF-1/CXCR4 激活NF-κB 信號傳導,NF-κB 被激活后可產生細胞周期蛋白D1 和MMP-13 。此外,該試驗表明SDF-1 激活p38 和Erk1/2 和NF-kB 信號通路,這些信號通路對軟骨細胞的增殖和成熟有不同的影響。其中,p38 MAP 激酶和NFkB 對SDF-1 介導的MMP-13 誘導至關重要,而MEK/Erk 途徑僅部分參與。相反,MEK/Erk 和NF-κB 在SDF-1 誘導細胞周期蛋白D1 中起關鍵作用,但p38 MAP 激酶途徑的作用有限[7]。
4 JAK/STAT 在OA 發展中的作用
ANTONIO 等[31]分析了JAK/STAT 途徑的SDF-1a 激活,發 現SDF-1 與CXCR4 結合 后,JAK2 和JAK3 與CXCR4 結合并且可通過轉磷酸作用以Gai 非依賴性方式激活,證明JAK2 和JAK3 都被激活并與CXCR4 相關,這種激活使得STAT 家族轉錄因子的幾個成員的募集和酪氨酸磷酸化成為可能。同時他們的研究表明JAK/STAT 途徑的激活是CC 和CXC 趨化因子的一般信號傳導途徑。JAK/STAT 信號傳導的激活對于使OA 進展為滑膜關節衰竭的慢性炎癥狀態永久化是至關重要的。該領域目前的研究揭示了在這些條件下調節異常細胞存活,細胞凋亡和基質金屬蛋白酶基因活性的分子機制[32]。
人和牛的軟骨細胞模型實驗中發現,JAK/STAT 在調節組織金屬蛋白酶組織抑制因子(tissue inhibitor of metalloproteinase,TIMP)和MMPs 中起重要作用,TMIP 在關節組織中廣泛表達,能夠抑制MMP,具有抑制軟骨在吸收的潛力[33],軟骨細胞通過JAK/STAT 調節MMP-TIMP 平衡,當信號通路受損,MMP-TIMP 失衡,MMP 表達過多,軟骨退化加速,關節炎便會進展加快[34]。見圖1。
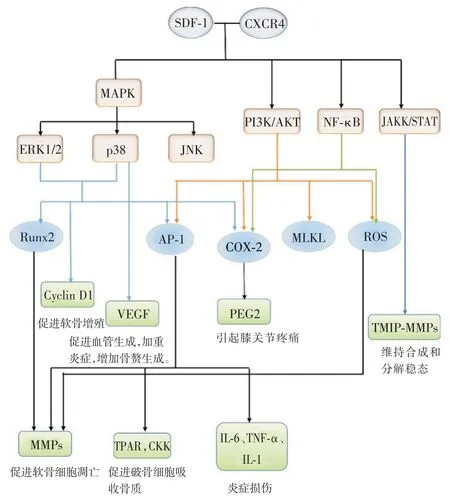
圖1 SDF-1/CXCR4 及其下游信號信號通路傳導圖Fig.1 The graph of signal path in SDF-1/CXCR4 and the downstream signal
CXCR4 結合激活的下游信號通路并不是一一對應的關系,而是相互交錯,互相影響。因此SDF-1與CXCR4 的結合后激活多種下游信號通路,涉及多種細胞因子,形成復雜的信號網絡,可誘導運動、趨化反應、粘附,以及分泌基質金屬蛋白酶、血管生成因子、白細胞介素、腫瘤壞死因子等,從而發揮生物學效應,調節細胞增殖,分化,存活和凋亡。SDF-1/CXCR4 信號通路與OA 中的各種病理事件有關。這些包括增加的炎癥,血管生成和骨和軟骨破壞,其參與風濕性疾病的發展。通過OA的體外和體內模型證明SDF-1 在這些相關過程中起關鍵作用。
5 總結與展望
鑒于SDF-1/CXCR4 信號通路在骨性關節炎的進展中具有重要作用,目前越來越多的學者將治療骨性關節炎的目光放在了阻斷SDF-1/CXCR4 信號通路上。因此,在已有幾種疾病的臨床試驗中正在研究阻斷CXCR4 受體的新治療劑的功效和安全性,AMD3100 和TN14003 等阻斷SDF-1/CXCR4通路的藥物目前正用于治療OA 的前期研究,這些藥物作為治療OA 的更為精準的靶向藥物提供了一種很有前景的治療方法。另外SDF-1/CXCR4 及其下游信號通路網絡是一個龐大的信息庫,與人體多種疾病相關,隨著研究的不斷深入,未來將會從組織、細胞、分子等層面系統深入研究SDF-1/CXCR4 信號通路調控骨性關節炎軟骨退變效果及分子機理,OA 的病因和機制也會越來越明晰,這對于無論是OA 的早期診斷或是治療都極大的進步,也為研發高效安全的骨性關節炎靶向治療藥物提供依據,從而使針對SDF-1/CXCR4 乃至其下游信號通路的拮抗劑將會有越來越多的發現,為臨床治療OA 提供全新的理念,豐富傳統的單一的手術治療OA 的方式。為延緩OA 進展提供新的思路,而OA 的治療也將隨著研究的進展更趨完善。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
