揮發性有機物與非甲烷總烴的相關性研究
2020-01-09 03:31:56邢利利張曉旭謝永洪吳曉峰
中國測試 2019年12期
李 佳,陳 勇,張 渝,邢利利,張曉旭,謝永洪,謝 翔,吳曉峰
(1.成都市環境監測中心站,四川 成都 610072; 2.四川省生態環境監測總站,四川 成都 610072)
0 引 言
揮發性有機物(volatile organic compounds,以下簡稱VOCs)是一類有機化合物質的統稱,世界衛生組織將其定義為“熔點低于室溫而沸點在50 ℃~260 ℃之間的揮發性有機化合物的總稱”[1-2]。由于VOCs是形成臭氧(O3)和細顆粒物(PM2.5)污染的重要前體物,且部分物質具有高毒性,生態環境部印發《2018年重點地區環境空氣揮發性有機物監測方案》加強對VOCs的監測,其中甚至將乙烷等沸點遠低于50 ℃的有機物也作為了VOCs進行監測。VOCs作為環境監測的重要指標,目前常用的監測方法主要有氣相色譜-質譜法(gas chromatographymass spectrometry,簡稱GC-MS)、氣相色譜-氫火焰離子化檢測器法(gas chromatography-hydrogen flame ionization detector,以下簡稱GC-FID)、氣相色譜-電子捕獲檢測器以及液相色譜法[3-5],我國針對大氣中VOCs多組分的監測主要有《環境空氣 揮發性有機物的測定 罐采樣/氣相色譜-質譜法》(HJ 759-2015)、《環境空氣 揮發性有機物的測定 吸附管采樣-熱脫附/氣相色譜-質譜法》(HJ 644-2013)等方法,但VOCs種類繁多、成分復雜,暫時還無法對所有組分進行監測。由于絕大部分VOCs在FID上都有不同靈敏度的響應,因此在環境標準中通常使用適用性受限的非甲烷總烴(non-methane hydrocarbon,以下簡稱NMHC)作為揮發性有機化合物總量的指標[3]。這影響了對VOCs總量核算的科學性和準確性,更忽視了不同組分在大氣化學中的作用差別,以及高危組分的濃度情況。
NMHC是指除甲烷氣體外,在氣相色譜儀的氫火焰離子化檢測器上有響應的氣態有機化合物的總和[6-8]。即在FID上有響應的碳氫化合物的衍生物也屬于NMHC范圍,在FID上沒有響應的有機物則不屬于非甲烷總烴,所以NMHC與VOCs在物質組成上有重疊,但不等同。另外,由于不同類型VOCs在FID上響應不同[9-11],所以NMHC與VOCs兩者在數值上不相等,但存在相關性。為研究NMHC與VOCs的數量關系,本文運用不同實驗方法所得的實驗結果建立了NMHC與VOCs的簡單數學模型,實現了VOCs與NMHC之間的數量轉換。
1 實驗部分
1.1 實驗原理與實驗儀器
實驗采用目前環境監測領域中3種不同原理的GC-FID方法對各類標準氣體和實際樣品進行NMHC的測定,3種方法分別是:六通進樣閥雙色譜柱法(以下簡稱雙柱法)、十通進樣閥單色譜柱法(以下簡稱單柱法)以及便攜式催化-GC-FID法(以下簡稱催化法)。
雙柱法:樣品經六通閥進樣,分別在甲烷柱和總烴柱上測得甲烷和總烴的含量,兩者之差即為NMHC,同時以除烴空氣代替樣品,測得氧在總烴柱上的響應值,以扣除樣品中氧對總烴測定的干擾[6-7]。檢測儀器為4890D氣相色譜儀(美國HP公司),其甲烷柱為內填充了粒徑為60~80目GDX-502的不銹鋼填充柱(2 m×3.2 mm),總烴柱為內填充了粒徑為60~80目硅烷化玻璃微珠的不銹鋼填充柱(2 m×3.2 mm)。儀器流路詳見圖1。
單柱法:樣品通過十通進樣閥進樣,由載氣將樣品帶入Porapak Q填充柱中分離,當氧氣和甲烷出峰后,通過閥的切換,其余組分一起反吹進入FID,形成一個NMHC合峰[12]。檢測儀器為Trace GC Utra氣相色譜儀(美國Thermo公司),其色譜柱為Porapak Q填充柱(美國Restek公司,2 m×3.2 mm)。儀器流路詳見圖2。
催化法:氣體樣品分別進入總烴檢測單元和甲烷催化轉換單元,經FID檢測器分別測定總烴及甲烷的含量,兩者之差即為NMHC的含量[13]。檢測儀器為PF-300便攜式甲烷/總烴/非甲烷總烴測試儀(意大利Pollution公司),儀器配有采樣系統、催化裝置和FID檢測器。
實際樣品中VOCs的測定:106種VOCs組分測定參考《環境空氣 揮發性有機物的測定 罐采樣/氣相色譜-質譜法》[14],用內壁惰性化處理的不銹鋼罐采集樣品,經預濃縮儀熱解析后,進入氣相色譜儀分離,用質譜檢測器進行定性與定量分析。檢測儀器為7200預濃縮儀(美國Entech公司),7890B-5977B氣相色譜質譜聯用儀(美國Agilent公司),其色譜柱為DB-624(美國Agilent公司,60 m×0.250 mm×1.4 μm);甲醛的監測方法為《空氣質量甲醛的測定 乙酰丙酮分光光度法》(GB/T 15516-1995)[15],檢測儀器為UV5500PC紫外可見分光光度計(上海元析儀器有限公司)。
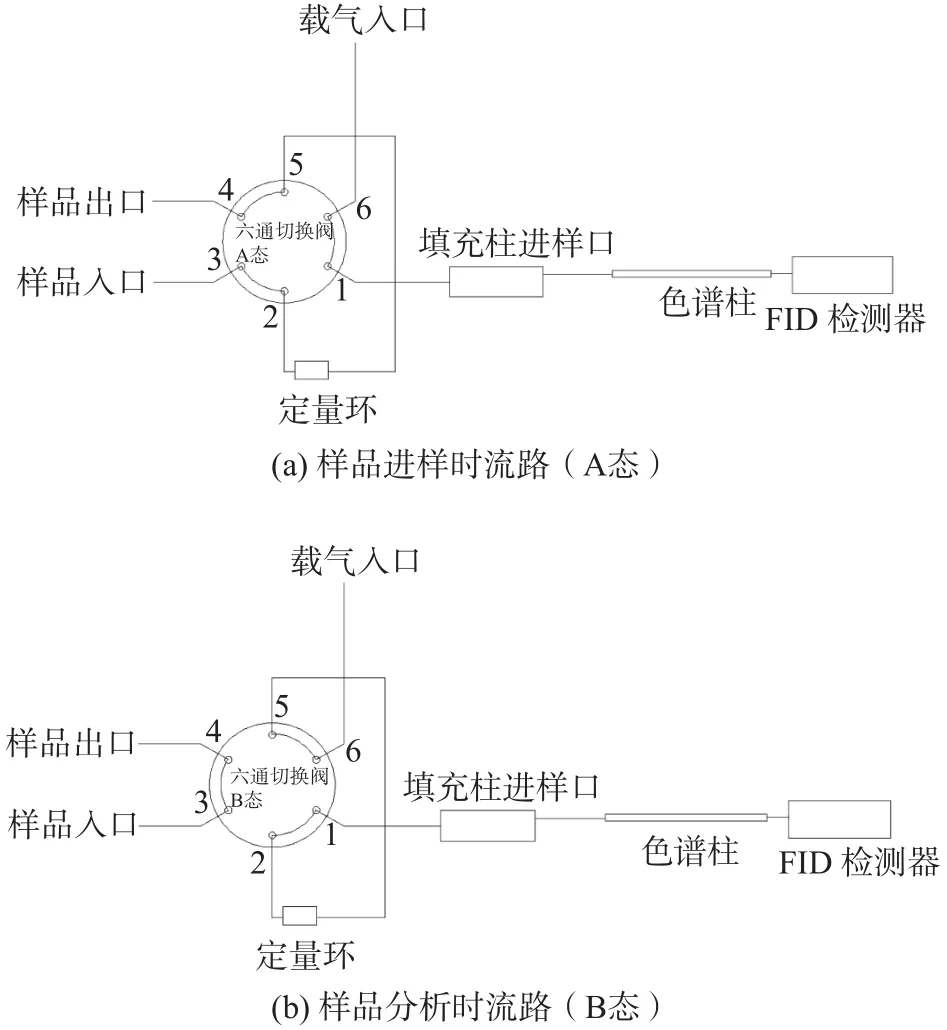
圖1 雙柱法儀器流路圖
標準液體樣品的測定:實驗采用液體標準樣品直接進樣,由載氣帶入色譜柱中分離,經FID檢測。檢測儀器為7890B(美國Agilent公司)氣相色譜儀,其色譜柱為BD-624毛細管柱(美國Agilent公司、30 m×0.25 mm×1.4 μm)。
1.2 實驗耗材
高純氮氣(≥99.999%),標準氣體1為內含66種組分的TO15(四川中測標物科技有限公司,140 mg/m3),標準氣體2為內含57種組分的PAMS(美國Restek公司,205 mg/m3),標準氣體3為內含5種組分的有機硫(四川中測標物科技有限公司,5.89 mg/m3),標準氣體4為內含兩種組分的醛類(四川中測標物科技有限公司,16.1 mg/m3),標準氣體5為內含兩種組分的NMHC(四川中測標物科技有限公司,甲烷為150 mg/m3,丙烷為296 mg/m3),標準氣體6為甲烷氣體(意大利Pollution公司,甲烷為40.0 mg/m3和120 mg/m3)。各類標準氣體濃度均以碳計,混合標準氣體濃度為各組分濃度之和。
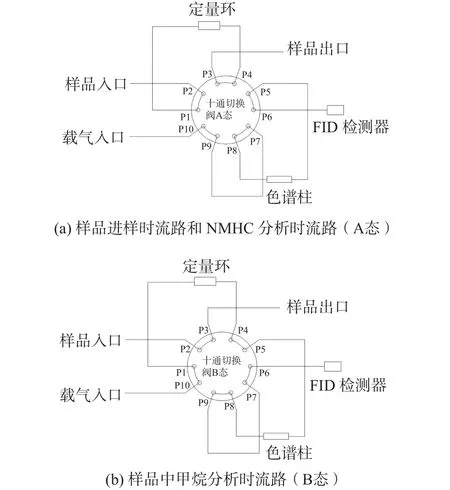
圖2 單柱法儀器流路圖
使用4700靜態稀釋儀(美國Entech公司)將各類標準氣體稀釋至硅烷化真空罐(美國Entech公司)中,得到不同濃度值(以碳計)樣品。標準氣體5配制成5個不同濃度值(甲烷濃度為:1.00 ,2.00 ,10.00 ,30.0 ,50.0mg/m3;總烴濃度為:3.00 ,6.00 ,30.0 ,90.0 ,150 mg/m3;NMHC濃度為:2.00 ,4.00 ,20.0 ,60.0 ,100 mg/m3);標準氣體1和標準氣體2配制成濃度60.0 mg/m3的樣品;標準氣體3和標準氣體4配制成濃度為4.00 mg/m3的樣品。以下若無特別說明,濃度值均以碳計。
1.3 實驗條件
雙柱法:進樣系統(包含進樣管路與六通閥)溫度20 ℃、65 ℃和150 ℃;進樣口恒壓30.0 kPa;進樣口溫度150 ℃;進樣量1.0 mL;檢測器250 ℃;氫氣壓力200 kPa;空氣壓力350 kPa;總烴柱溫150 ℃、甲烷柱溫100 ℃。
單柱法:進樣系統(進樣管道與十通閥)溫度100 ℃;柱箱100 ℃保持4.0 min;載氣30.0 mL/min、氫氣40.0 mL/min、空氣300 mL/min;檢測器溫度250 ℃;十通閥切換時間分別是0.01 min和0.60 min。
催化法:采樣流量800 mL/min;進樣口溫度180 ℃;檢測器溫度190 ℃;催化模塊溫度280 ℃;點火階段氫氣流量30.0 mL/min、空氣流量300 mL/min;樣品測定階段氫氣流量:25.0 mL/min、空氣流量:225 mL/min。
標準液體樣品的測定:進樣口溫度220 ℃;柱流量1.5 mL/min;柱溫35 ℃保持6 min,10.0 ℃/min升溫至220 ℃保持6 min;檢測器溫度220 ℃;空氣流量400 mL/min;氫氣流量30.0 mL/min。
2 結果與討論
2.1 色譜圖與校準曲線
2.1.1 色譜圖
除催化法無色譜柱外,雙柱法和單柱法的色譜圖如圖3、圖4所示。其中圖4中兩個未標注的色譜峰為十通閥兩次切換形成的雜峰。
2.1.2 校準曲線
除催化法使用甲烷標準氣對儀器進行單點校準外,雙柱法與單柱法均使用NMHC標準氣體的濃度與峰面積繪制了校準曲線,其相關系數均大于0.999。校準曲線與相關系數見表1。

圖3 雙柱法色譜圖

圖4 單柱法色譜圖

表1 不同方法的校準曲線與相關系數匯總表
2.2 實驗條件的優化
雙柱法測定樣品時,由于VOCs組分復雜,可能存在強吸附性、較高沸點的物質,在進樣流路中易因吸附冷凝造成損失,最常見的解決辦法是提高進樣系統的溫度。由于烴類物質在FID上的響應與其含碳數呈現正相關[8-11],本實驗選用PAMS標準氣體(為C2~C12的烴類,包括了較高沸點的VOCs組分)考察進樣系統的溫度對實驗結果的影響,便于判斷損失情況。綜上,為考察進樣系統溫度對測定結果的影響,實驗采用雙柱法分別在進樣系統為20 ℃(常溫)、65 ℃和150 ℃(接近部分廠家切換閥的耐受上限溫度)時測定PAMS標準氣體樣品。實驗時每個樣品均平行測定了3次(n=3)并取均值,將其除以理論值得到回收率。
不同進樣系統溫度測定PAMS標準氣體結果如圖5。實驗測定值隨著進樣系統溫度的升高而增加,原因是VOCs各組分在進樣過程中有不同程度的吸附,溫度升高可以有效地降低吸附,當溫度升高至150 ℃后,吸附現象基本消失,測定值與理論值基本一致。因此,本文的后續實驗,進樣系統的溫度均設置為150 ℃。
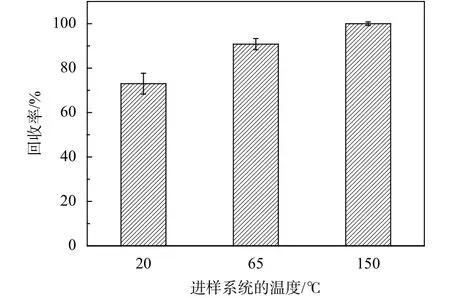
圖5 不同進樣系統溫度測定PAMS標準氣體結果匯總圖
單柱法測定標準氣體樣品時,由于Porapak Q對組分的保留能力與其極性、分子量呈正相關,分析含有強極性和較高分子量組分的樣品時,柱箱溫度可能影響結果的準確性。為考察柱箱溫度對實驗結果的影響,實驗時分別在柱箱溫度為80 ℃、100 ℃、140 ℃和160 ℃時測定PAMS標準氣體樣品。如圖6所示,圖中兩個未標注的色譜峰為十通閥兩次切換形成的雜峰。

圖6 不同柱溫下測定PAMS標準氣體色譜圖
不同柱箱溫度下測定PAMS所得色譜圖如圖6。若柱箱溫度較低,極性或沸點達到一定水平的組分很難出峰,若升高柱箱溫度,儀器基線會隨溫度升高而急劇上升,基線對色譜峰的掩蓋造成無法對色譜峰進行有效積分。綜上,實驗選擇柱箱溫度100 ℃作為最佳實驗條件,但是此實驗條件NHMC色譜峰依然有不同程度的拖尾現象,自動積分會有較大偏差,結果處理時采用了手動積分,手動積分結果RSD≤3.5%。
2.3 標準氣體測定結果
3種方法測定各類標準氣體的回收率(測定值除以理論值)詳見圖7。實驗結果表明:1)雙柱法、單柱法測定各類標準氣體樣品呈現很好的一致性;2)催化法測定PAMS和TO15的回收率均較低,并且在測定TO15時,儀器顯示甲烷的測定值為4.72 mg/m3,而TO15標準氣體組分中并沒有甲烷,甲烷理論上應為未檢出。原因是催化劑對VOCs各組分的催化效率不同,導致樣品并沒有完全轉變為CO2和H2O,所以,當分析難催化分解物質時,催化劑的催化效率可能會影響實驗結果的準確性。3)PAMS回收率為100%,但TO15、有機硫與醛類的測定值均低于理論值,回收率最低為醛類。原因是PAMS組分為烷烴、烯烴和芳香烴類物質,而TO15組分為烷烴、芳香烴、鹵代烴以及其他含氧化合物,有機硫組分為二甲基二硫醚、乙硫醚、甲硫醚、乙硫醇和甲硫醇,醛類組分為甲醛和乙醛,物質中含非碳氫原子使其在FID上的響應與同數碳烷烴類物質的響應不同,正如盛立彥[16]等人提出關于醇醚類物質在FID上響應會降低一樣,因含氧原子增加了碳正離子形成的難度,從而降低了分子的有效碳數,使得物質在FID上的響應降低。本文標準氣體樣品的測定結果也正好驗證了不同類型VOCs在FID上的摩爾響應值存在差異[16-18]。為考察不同類型VOCs在FID上響應的差異性,實驗采用液體標樣直接進樣、GC-FID分析的方式,考察單一組分在FID上的響應。
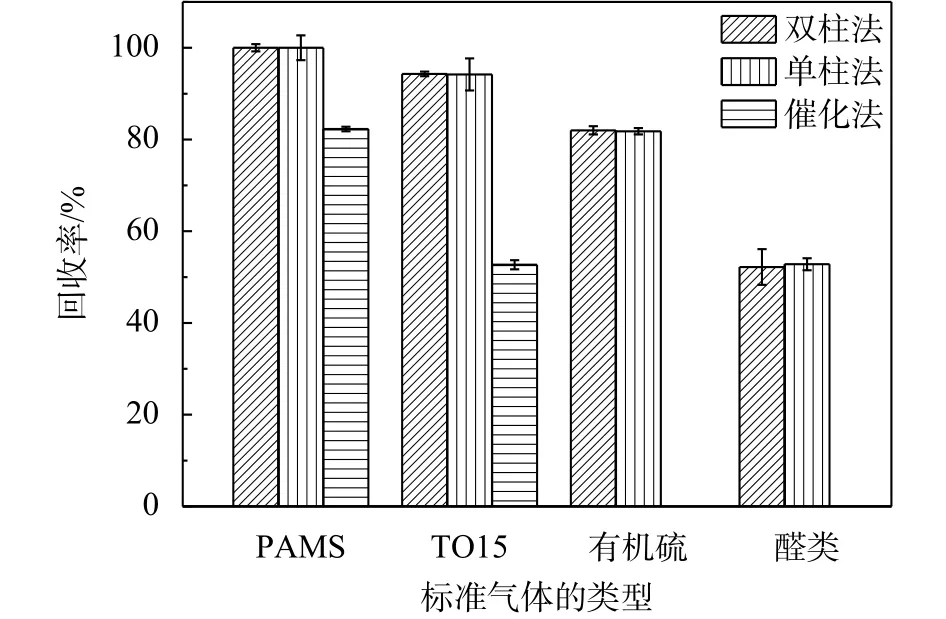
圖7 3種方法測定標準氣體結果匯總圖
2.4 VOCs單一組分在FID上的響應以及VOCs與NMHC的相關性
由FID原理可知,當物質進入FID燃燒的氫氣火焰中,與氧氣進行了化學電離反應。以苯為例,其在FID的反應如下[9]:
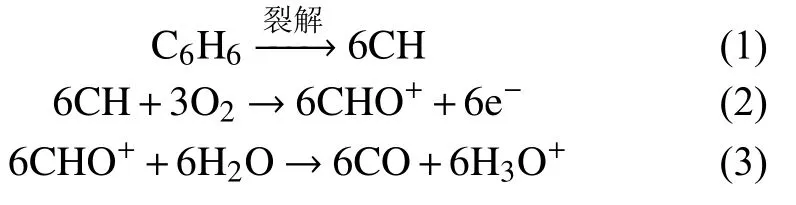
反應表明,電流產生的正離子和電子形成了微電流信號經電流放大器放大后,由儀器記錄儀畫出色譜峰。故本文使用等質量組分、且VOCs類別包含烷烴類、烯烴類、芳香烴類、酮類、醇類、酯類以及鹵代類有機化合物的3種液體標樣分別直接進樣后經色譜柱分離,由FID檢測器檢測得到單一組分在FID上的色譜峰面積,如圖8所示。
GC-FID法測定等質量VOCs標準液體結果詳見圖8。比較每一種組分在FID上的響應可以發現:只含碳氫的分子在FID上的響應與碳原子數量呈現一比一正比關系;含有極性共價鍵的分子在(1)反應中,若碳原子與非碳原子的共價鍵斷裂形成的碳原子基團傾向于失去電子,將會抑制反應式(2)的進行,物質在FID上的響應減弱;相反,若傾向于得到電子,將會促進反應式(2)的進行,物質在FID上的響應增強。
物質在FID上的響應值最終以峰面積呈現,若用A表示某一種物質在FID上的總有效響應值,那么:
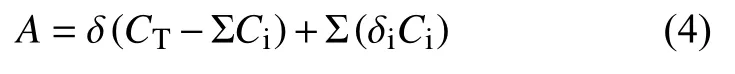
式中:δ——摩爾碳的有效響應值;
CT——物質的總碳數;
Ci——極性共價鍵的碳原子;
δi——極性共價鍵的碳原子摩爾響應值;
D——極性共價鍵的電負性,其中δi=f(δ,D)。
理論上可通過極性共價鍵的電負性D算得所有物質的A,即當沒有標準物質的情況下,也能夠通過其他物質的響應來推算無標待測組分的濃度。但實際上受實驗條件的限制,本文難以準確測定所有極性共價鍵的電負性,故建立了簡化的響應計算數學模型:將物質中碳碳相連的碳原子視為摩爾碳響應值均為δ,根據本文GC-FID測定VOCs標準液體的實驗結果,以FID上碳原子離子化效率為100%的苯為基準,計算得到碳原子在FID上的摩爾碳響應值δ,與非碳原子相連的碳原子視為摩爾碳響應值為δi,假設δi與δ的比值為di,結合在本文實驗條件下得到的標準氣/液體在FID上的響應值,對不同官能團的δi計算,根據δ與δi可以對di值進行估算,以此建立VOCs與NMHC的簡單數學模型。上段的公式則可以轉變為:A=δ∑(diCs)=δd表征,公式中Cs為物質含的碳元素,對于只與碳氫原子相連的Cs,其對應di值為1,其他部分官能團的di值與Ci值見表2。

圖8 GC-FID法測定標準液體結果匯總圖(等質量響應值)

表2 部分官能團的di值與Ci值匯總表
取代官能團會抑制或促進反應式(2),從而造成物質在FID上的響應降低會增加,但根據結果分析,取代官能團難以完全的抑制或無限的促進有機分子在FID上的響應,所以根據實驗結果將模型方程式擬合為較接近實際情況的指數函數或冪函數。
2.5 標準氣體結果分析
運用數學模型對各類標準氣體的理論響應值進行修正,得到修正理論值,計算雙柱法的測定值與理論修正值的百分比值(修正回收率),同時將標準氣體測定值與理論值計算所得百分比值(實測回收率)進行比較。
雙柱法測定各類標準氣體的實測回收率與修正回收率的比較詳見圖9。如圖,通過數學模型的修正后,各類標準氣體回收率均達到了98.1%~100%。
2.6 實際樣品結果分析
實驗使用單柱法和雙柱法測定了實際樣品的NMHC,同時使用預冷濃縮/GC-MSD測定實際樣品中106種有機組分(主要有烷烴、烯烴、芳烴和少量醇、酮類),以及使用乙酰丙酮分光光度法測定樣品中甲醛的含量(106種組分中不包括甲醛,而甲醛是室內重要有機物指標)。
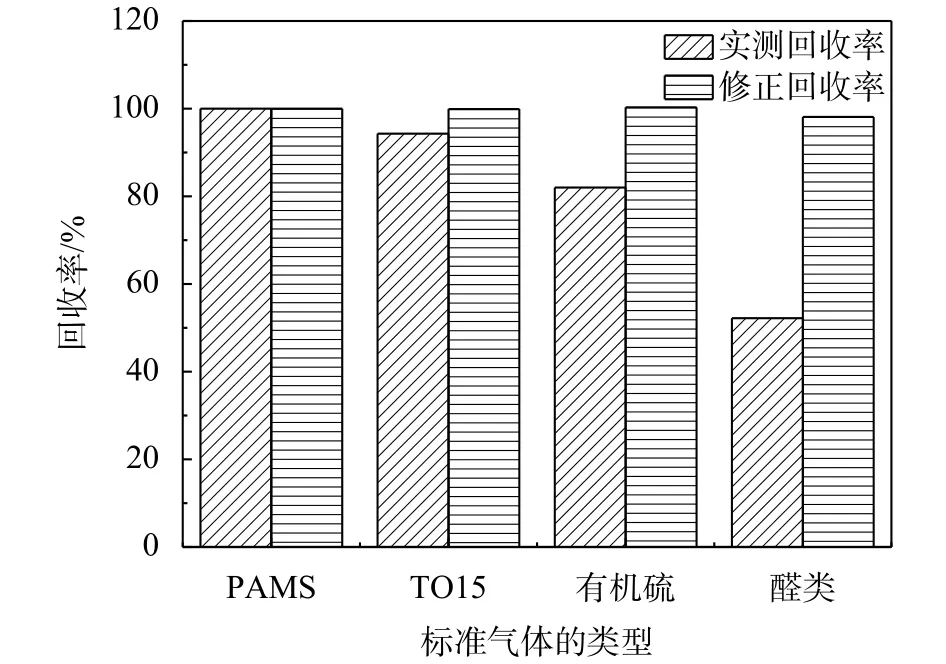
圖9 各類標準氣體實測回收率與修正回收率比較圖
實際樣品的NMHC與VOCs(包括甲醛在內的107種有機物組分的質量體積濃度之和)的測定結果詳見圖10。實驗結果比較發現:樣品NMHC的測定值高時,VOCs的測定值也普遍較高,兩者存在一定程度的正相關關系;由于實際樣品中VOCs的種類繁多,而實驗室能檢測的種類有限,所以VOCs的各組分濃度之和低于NMHC。
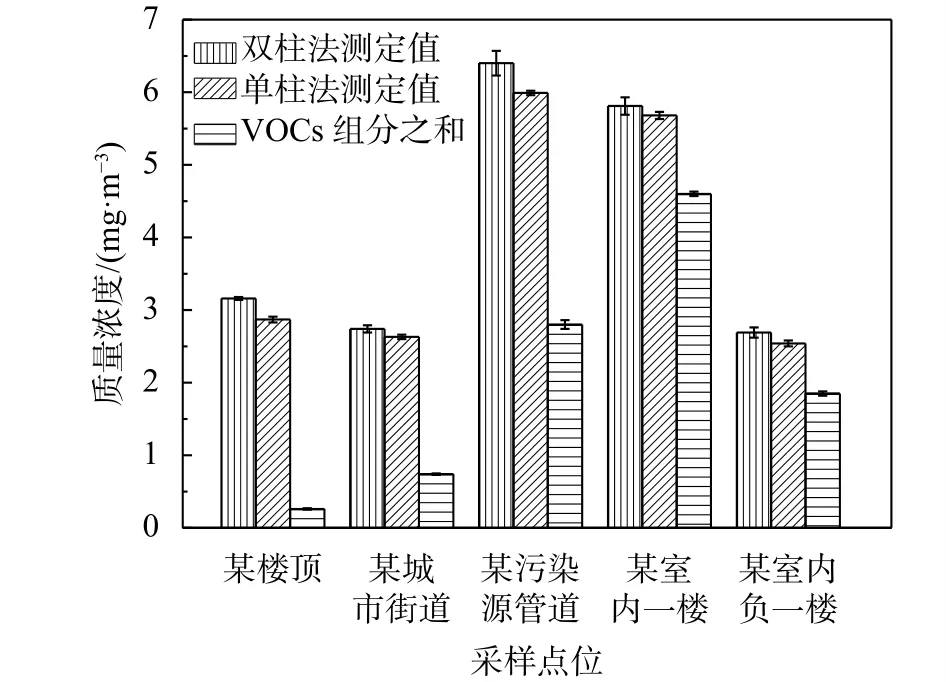
圖10 實際樣品NMHC測定值與VOCs結果比較圖
3 結束語
綜上,VOCs與NMHC之間有相關性,但并不等同。從物質組成上,NMHC與VOCs有重疊,但不完全相同。從數值上,雙柱法、單柱法和催化法3種實驗方法所得測定值已表明NMHC與VOCs兩者之間存在一定的數量關聯:當樣品中只含碳氫化合物時(如PAMS和苯系物),VOCs與NMHC的數值十分接近,此時可以用NMHC作為VOCs的總量指標;當VOCs分子含有取代官能團時,其在FID上的響應系數與碳氫化合物的響應系數不同,NMHC與VOCs的數值有較大差異,此時用NMHC將難以對VOCs總量做出科學準確的評估。
此外,NMHC僅為總量型的指標,而且對含官能團的VOCs分子響應一般偏低,因此無法對甲醛、1,1,1-三氯乙烷、四氯化碳等高毒VOCs物質的濃度進行有效監測或評估,存在環境監測方面難以彌補的缺陷。
本文結合標準氣體與標準液體的在FID上的測定結果建立了NMHC與VOCs之間的簡單數學模型,且運用該模型計算得出各類標準氣體的VOCs理論修正值,NMHC的回收率為98.1%~100%,數學模型可信。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
專用汽車(2016年4期)2016-03-01 04:13:43
