代謝工程改造大腸桿菌合成反式-4-羥基-L-脯氨酸
2020-02-10 15:35:16韓亞昆吳鶴云徐慶陽謝希賢
食品科學 2020年2期
關鍵詞:產量
李 強,韓亞昆,蔣 帥,張 悅,吳鶴云,徐慶陽,謝希賢
(天津科技大學生物工程學院,代謝控制發酵技術國家地方聯合工程實驗室,天津 300457)
反式-4-羥基-L-脯氨酸是一種稀有亞氨基酸[1],主要以多肽形式存在于動物的膠原蛋白中[2]。反式-4-羥基-L-脯氨酸具有延緩衰老功效以及抗腫瘤、抗高血壓作用[3-4],也被用于合成碳青霉烯類抗生和消炎藥等藥物[5-6],因在醫藥保健、材料化工、食品營養和護膚美容等行業都具有廣泛應用[7]。近年來,隨著反式-4-羥基-L-脯氨酸在醫藥和美容領域應用的顯著增加,其市場需求量逐年增加,這對反式-4-羥基-L-脯氨酸的工業生產提出了更高的要求。
反式-4-羥基-L-脯氨酸的生產方法主要包括蛋白質水解法、化學合成法和微生物發酵法[8]。蛋白質水解法和化學合成法工藝復雜、原料成本高,而微生物發酵法具有周期短、易控制、產率高和污染小等優點,是大規模工業化生產反式-4-羥基-L-脯氨酸的理想方法。20世紀60年代開始,國內外很多科學家就開始對反式-4-羥基-L-脯氨酸生物合成開展研究,主要集中在L-脯氨酸-4-羥基化酶篩選和反式-4-羥基-L-脯氨酸產生菌構建方面。Baldwin等[9]首次從Streptomyces grixaviridru P8648分離得到L-脯氨酸-4-羥化酶,并確定了該酶的底物特異性。Shibasaki等[10]從指孢囊菌RH1中鑒定了L-脯氨酸-4-羥基化酶的基因,隨后將該基因克隆轉入大腸桿菌W1485,該重組菌在添加前體物L-脯氨酸的條件下發酵100 h,反式-4-羥基-L-脯氨酸產量達到41 g/L,生產強度達到0.41 g/(Lg h)。后,Shibasaki等[11]進一步將密碼子優化后的指孢囊菌的L-脯氨酸-4-羥基化酶基因引入L-脯氨酸生產菌中,以葡萄糖為底物發酵99 h,反式-4-羥基-L-脯氨酸產量達到25 g/L,生產強度達到 0.25 g/(Lg h),為直接發酵法生產反式-4-羥基-L-脯氨酸奠定了基礎。為構建發酵性能更好的反式-4-羥基-L-脯氨酸的生產菌株,Falcioni等[12]嘗試在谷氨酸棒狀桿菌來源的L-脯氨酸生產菌引入密碼子優化后的指孢囊菌 L-脯氨酸-4-羥基化酶基因,發酵23 h,反式-4-羥基-L-脯氨酸產量達到7.1 g/L,生產強度達到0.31 g/(Lg h)。林凡[13]在Escherichia coli BL21(DE3)導入含密碼子優化后的指孢囊菌的L-脯氨酸-4-羥基化酶基因,發酵64 h,反式-4-羥基-L-脯氨酸產量達到41.5 g/L,生產強度達到 0.65 g/(Lg h),這也是目前報道發酵水平最高的菌株。這些工程菌的構建和改進推動了反式-4-羥基-L-脯氨酸的微生物發酵法生產。

圖 1 大腸桿菌反式-4-羥基-L-脯氨酸合成代謝路徑Fig. 1 Biosynthetic pathway of trans-4-hydroxy-L-proline in E. coli
如前所述,雖然已有一些反式-4-羥基-L-脯氨酸工程菌的構建研究,但現有菌株的生產效率仍然偏低,這也導致了反式-4-羥基-L-脯氨酸的生產成本偏高。另外,由于這些菌株通過質粒克隆的方式引入L-脯氨酸-4-羥基化酶基因,一方面增加了菌體的負擔,另一方面會出現因質粒丟失導致發酵不穩定,這些也限制了菌種在規模化生產中的應用。針對這些問題,本研究從Micromonospora sp. CNB394篩選到一個高活性的L-脯氨酸-4-羥基化酶,并采用系統代謝工程技術通過阻斷L-脯氨酸的降解、解除L-脯氨酸反饋抑制、強化L-脯氨酸合成途徑,以及引入羥化反應途徑,構建高效率的反式-4-羥基-L-脯氨酸工程菌(圖1)。
1 材料與方法
1.1 材料與試劑
1.1.1 菌株與質粒
本研究所使用和構建的菌株、質粒見表1。

表 1 實驗所用菌株和質粒Table 1 Strains and plasmids used in this study
1.1.2 試劑
Primer STAR HS DNA聚合酶 大連寶生物科技有限公司;2h Rapid Taq Mix、ClonExpress?II One Step Cloning Kit 南京諾唯贊生物科技有限公司;質粒快速提取試劑盒、DNA凝膠純化回收試劑盒 美國Omega 公司;引物及基因均由蘇州金唯智科技有限公司合成。
1.2 儀器與設備
5 L發酵罐 上海保興生物設備有限公司;S-433D氨基酸分析儀 北京捷盛依科科技有限公司;SBA-40C生物傳感儀 山東省科學院生物研究所。
1.3 方法
1.3.1 工程菌構建
菌株構建過程中,利用規律成簇的間隔短回文重復-associated protein 9(clustered regularly interspaced short palindromic repeats-associated protein 9,CRISPRC a s 9)基因編輯方法進行基因的敲除和整合[14]。CRISPR-Cas9系統包括pGRB和pREDCas9兩個質粒,其 中p R E D C a s 9 攜 帶g R N A 表 達 質 粒p G R B 的消 除 系 統、λ 噬 菌 體 的R e d 重 組 系 統[15]及C a s 9 蛋白表達系統[16],含奇霉抗性(工作質量濃度 100 mg/L),32 ℃培養。pGRB以pUC18為骨架,包括啟動子J23100、gRNA-Cas9結合區域序列和終止子序列,含氨芐青霉抗性(工作質量濃度100 mg/L),37 ℃培養。構建質粒pGRB的目的是轉錄相應gRNA,從而與Cas9蛋白形成復合體,并通過堿基配對和PAM識別目的基因靶位點,實現目的DNA雙鏈斷裂。
1.3.1.1 gRNA質粒和重組DNA片段構建
為構建gRNA質粒,先設計合成兩條完全反向互補的單鏈DNA,然后通過退火形成雙鏈DNA,該雙鏈DNA中間序列是靶位點的特定gRNA間隔序列,兩端序列與pGRB具有同源序列。利用ClonExpress?II One Step Cloning Kit,雙鏈DNA與線性化pGRB通過同源重組形成gRNA表達質粒。利用引物設計軟件primer5,以待敲除基因或待整合位點的上下游序列為模板,設計上下游同源臂引物(擴增長度約400~500)。以待整合基因為模板,設計整合基因的擴增引物,聚合酶鏈式反應(polymerase chain reaction,PCR)擴增上下游同源臂和目的基因片段后,再經過重疊PCR制備重組片段。
1.3.1.2 DNA片段重組
首先,將pREDCas9質粒通過化學轉化的方式導入E. coli W3110感受態細胞中[17]。通過菌落PCR篩選正確的陽性重組子,菌株接種于LB培養基32 ℃過夜培養,然后按1%的接種量轉移到100 mL 2h YT培養基中(1.6%蛋白胨、1%酵母浸粉、0.5% NaCl)。32 ℃培養至細胞OD600nm達到0.1~0.2后,添加0.1 mmol/L異丙基-β-D-硫代半乳糖苷誘導重組酶表達。繼續培養至細胞OD600nm達到0.4~0.5,收集菌體制備電轉感受態細胞[18]。將200 ng重組DNA片段和100 ng gRNA質粒加入感受態細胞,并轉移至0.1 cm電轉杯中,在1 850 kV條件下進行電轉化[19]。 電轉化后的細胞加入1 mL SOC培養基中32 ℃復蘇2 h,然后取100~200 μL涂布到含氨芐和奇霉抗性的LB平板上。32 ℃過夜培養,挑取單菌落進行菌落PCR驗證,篩選陽性重組菌株。將陽性菌株在含有0.2%阿拉伯糖的LB培養基中培養,誘導pREDCas9中質粒消除系統切割pGRB。最后將菌株在42 ℃培養過夜,消除溫敏性質粒pREDCas9,獲得無質粒的工程菌株。
1.3.2 搖瓶發酵
1.3.3 發酵罐分批補料發酵
發酵罐的培養基與搖瓶發酵培養基相同。利用無菌水將茄形瓶斜面菌種制成菌懸液,接入總裝液量為3 L的5 L發酵罐中。通過pH值電極控制自動流加氨水(體積分數25%)維持pH 7.0左右;發酵過程溫度恒定在37 ℃;通過調節攪拌槳轉速和通風量控制溶氧在25%~35%之間。培養至細胞OD值達到12~16時,以15%接種量接入新鮮的發酵培養基。發酵過程中控制pH 7.0左右,溫度維持在37 ℃,溶氧維持在25%~35%之間。當培養基中的葡萄糖消耗完之后,以一定的速率流加80%的葡萄糖溶液,維持發酵培養基中的葡萄糖質量濃度在5 g/L以下[22]。
1.3.4 發酵過程中檢測與分析
大腸桿菌生物量利用紫外分光光度計在600 nm波長處的OD值檢測;發酵液中葡萄糖含量由SBA-40C生物傳感儀檢測;發酵液中谷氨酸、L-脯氨酸以及反式-4-羥基-L-脯氨酸的含量采用S-433D氨基酸分析儀檢測;氨基酸含量采用茚三酮柱后衍生法利用S-433D氨基酸分析儀測定[23]。色柱分析條件:色柱LCA K06/Na,進量50 μL,檢測波長570 nm和440 nm,流動相流速 0.45 mL/min,反應器溫度130℃。其中,L-脯氨酸和反式-4-羥基-L-脯氨酸反應后產物可在440 nm波長處被檢測到,其他氨基酸反應后產物可在570 nm波長處被檢測到。
1.4 數據統計學分析
發酵數據代表3 組平行發酵數據的f s。利用t檢驗雙尾分布對兩組發酵參數進行單向方差分析。P<0.05,差異顯著;P<0.01,差異極顯著。
2 結果與分析
2.1 木糖誘導基因表達系統建立
在代謝工程研究中,對相關的內源基因和異源基因進行過表達是常用的改造策略。T7啟動子是大腸桿菌中報道最強的啟動子,但是大腸桿菌自身沒有T7 RNA聚合酶,因首先需要在大腸桿菌中引入來源于T7噬菌體的RNA聚合酶[24]。另外,T7啟動子表達基因過強時,有時會影響菌體的生長,因需要控制T7 RNA聚合酶的表達時間及表達量。本研究首先利用CRISPR-Cas9基因編輯方法,以大腸桿菌W3110為出發菌,在lacZ位點整合了木糖啟動子控制的T7 RNA聚合酶基因,可實現在木糖誘導的條件下啟動T7 RNA聚合酶的表達[21]。隨后對dgsA(編碼糖代謝轉錄抑制因子)啟動子-10區進行點突變,解除了葡萄糖對木糖啟動子轉錄的阻遏作用[25]。最后對xlyA(木糖異構酶)進行基因敲除,阻斷木糖的代謝途徑,減少木糖因分解代謝導致的損失[26]。通過以上基因操作,構建的菌株HYP01可在木糖誘導的情況下實現相關基因的過表達。
2.2 強化L-脯氨酸合成途徑
2.2.1 L-脯氨酸操縱子過表達對L-脯氨酸合成的影響
γ-谷氨酸激酶為L-脯氨酸合成的限速酶,受到L-脯氨酸的反饋抑制。根據報道[27],將編碼γ-谷氨酸激酶基因proB中319位G替換成A,編碼蛋白的第107位氨基酸天冬酰胺替換為L-脯氨酸,可解除L-脯氨酸的反饋抑制。啟動子是代謝工程中調節基因表達的重要元件[28],其中T7啟動子(PT7)和trc啟動子(Ptrc)是大腸桿菌中應用最廣泛的兩種強啟動子[29],但兩者轉錄強度不同。本研究首先敲除了菌株HYP01 L-脯氨酸脫氫酶基因putA,阻斷前體物L-脯氨酸的降解途徑,構建菌株HYP02(表1)。
為驗證PT7和Ptrc對L-脯氨酸操縱子的表達效果,在菌株HYP02的假基因位點yghX處整合proB*A,分別由PT7和Ptrc啟動轉錄,分別構建了菌株HYP03和HYP04。搖瓶發酵結果如圖2所示,HYP03的L-脯氨酸產量達到5.5 g/L,HYP04的L-脯氨酸產量達到11.8 g/L,是HYP03 L-脯氨酸產量的2.14 倍。圖2表明,采用Ptrc控制L-脯氨酸操縱子的過表達,效果更好。

圖 2 不同啟動子控制L-脯氨酸操縱子過量表達的發酵參數比較Fig. 2 Comparison of fermentation parameters for overexpression of proline operon controlled by different promoters
氨基酸分析結果還顯示,HYP03和HYP04的發酵液中都有少量L-谷氨酸積累(圖2),說明L-谷氨酸到L-脯氨酸的代謝通量仍需要加強。由于L-脯氨酸操縱子在Ptrc控制下效果更好,本研究在菌株HYP04的假基因位點trpR處整合第2個拷貝的Ptrc-proB*A,構建了菌株HYP05。搖瓶發酵結果如圖3所示,HYP05的L-脯氨酸產量達到13.2 g/L,相比菌株HYP04(12.1 g/L)提高了9.1%,L-谷氨酸質量濃度為0.4 g/L,比HYP04降低了77.8%,說明L-脯氨酸操縱子基因雙拷貝進一步增強了 L-脯氨酸的合成通量。

圖 3 雙拷貝proB*A對L-脯氨酸產量的影響Fig. 3 Effect of double proB*A copy on proline production
2.2.2 增加胞內NADPH供應對L-脯氨酸合成的影響
NADPH是細胞內重要的還原力,對于多種氨基酸的合成代謝都具有重要的作用[30],氨基酸的生物合成往往因NADPH的供應不足而受到限制。如圖1所示,合成1 mol的L-脯氨酸需要3 mol NADPH,NADPH的供應也是L-脯氨酸合成的限制性因。大腸桿菌的基因pntAB編碼吡啶核苷酸轉氫酶PntAB,可以催化NADP(H)與NAD(H)之間氫負離子可逆轉移,增強PntAB的表達被證明能夠有效提高大腸桿菌胞內NADPH的供應[31]。由于PntAB是膜蛋白,表達量過大有時也會對菌體生長造成負面影響。本研究在HYP05的假基因yeeP位點處整合了自身啟動子控制的pntAB基因,構建了菌株HYP06。搖瓶發酵結果如圖4所示,HYP06的L-脯氨酸產量達到15.3 g/L,相比HYP05(13.1 g/L)提高了16.8%,說明增加NADPH的供應顯著提高L-脯氨酸的積累。

圖 4 增強NADPH供應對L-脯氨酸產量的影響Fig. 4 Effect of enhancing NADHP supply on proline production
2.3 高活性L-脯氨酸-4-羥基化酶的篩選
大腸桿菌中不存在L-脯氨酸-4-羥基化酶基因,反式-4-羥基-L-脯氨酸合成途徑的重構需要引入外源的 L-脯氨酸-4-羥基化酶基因,因高活性的L-脯氨酸-4-羥基化酶是反式-4-羥基-L-脯氨酸工程菌構建的關鍵。根據指孢囊菌RH1的L-脯氨酸-4-羥基化酶的氨基酸序列(GenBank:BAA20094.1),在NCBI數據庫通過BLAST比對,挑選了與L-脯氨酸-4-羥基化酶具有保守催化結構域的4 個基因(分別命名為p4H1、p4H2、p4H3和p4H4),分別來源于Micromonospora sp. CNB394、Nocardia sp. BMG111209、Kutzneria albida DSM 43870、Streptomyces sp. SA15。將密碼子優化后的p4h、p4H1、p4H2、p4H3和p4H4連接到pTrc99a,導入菌株HYP06中,構建了菌株HYP07、HYP08、HYP09、HYP10和HYP11。搖瓶發酵結果如圖5所示,HYP07、HYP08、HYP09和HYP10發酵液中反式-4-羥基-L-脯氨酸質量濃度分別為8.6、9.5、7.6 g/L和2.3 g/L,表明p4H1、p4H2、p4H3具有L-脯氨酸-4-羥基化酶的活性。來源于Micromonospora sp. CNB394的p4H1活性最高,與菌株HYP07相比,HYP08的反式-4-羥基-L-脯氨酸產量提高了10.5%。

圖 5 含L-脯氨酸-4-羥基化酶的菌株發酵參數比較Fig. 5 Comparison of fermentation parameters for recombinant strains with different L-proline-4-hydroxylases
2.4 L-脯氨酸-4-羥基化酶過表達對反式-4-羥基-L-脯氨酸合成的影響
為驗證p4H1在基因組上的表達活性,將p4H1整合于菌株HYP06的假基因位點tehB處,分別由PT7和Ptrc啟動轉錄,構建了菌株HYP12和HYP13。搖瓶發酵結果如圖6所示,HYP12的反式-4-羥基-L-脯氨酸產量達到4.9 g/L,比菌株HYP13(3.2 g/L)高53.1%;說明p4H1在PT7控制下的表達效果更好。
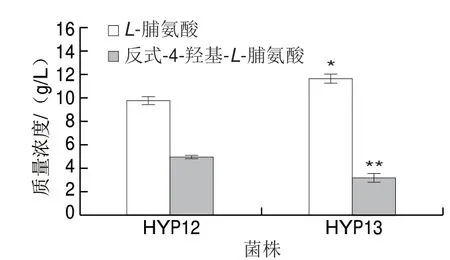
圖 6 不同啟動子控制p4H1過量表達的發酵參數比較Fig. 6 Comparison of fermentation parameters for overexpression of p4H1 controlled by different promoters
從圖6可知,HYP12和HYP13的發酵液中均有前體物L-脯氨酸殘留,說明L-脯氨酸-4-羥基化酶的表達量不足。為進一步提高反式-4-羥基-L-脯氨酸的產量,在HYP12假基因ycjV處整合第2個拷貝的PT7-p4H1,構建了菌株HYP14。如圖7所示,HYP14的反式-4-羥基-L-脯氨酸產量達到11.7 g/L,相比HYP12(5.0 g/L)提高了 1.34 倍,L-脯氨酸質量濃度降低到3.5 g/L。隨后在HYP14的假基因yjiP位點進行整合了第3個拷貝的PT7-p4H1,構建了菌株HYP15。反式-4-羥基-L-脯氨酸產量達到13.6 g/L,相比HYP14提高了16.2%,L-脯氨酸質量濃度降低到1.5 g/L。

圖 7 p4H1拷貝數對發酵參數的影響Fig. 7 Effect of p4H1 copy number on fermentation parameters
2.5 工程菌發酵罐分批補料發酵
為評估菌株HYP15的生產性能,在5 L發酵罐中進行分批補料發酵實驗。如圖8所示,隨著發酵時間延長,細胞生物量逐步增加,最終OD600nm達到了70。12~36 h,反式-4-羥基-L-脯氨酸保持了較高的合成速率,40 h的產量達到48.6 g/L,糖酸轉化率(最終合成反式-4-羥基-L-脯氨酸總量與上發酵消耗總糖量之比)和生產強度分別達到了21.6%和1.22 g/(Lg h)。發酵液中可檢測到的副產物有L-脯氨酸和乙酸,質量濃度分別為2.3 g/L和0.9 g/L。
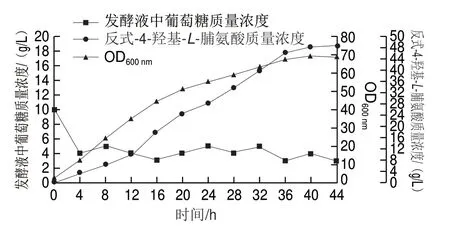
圖 8 工程菌HYP15在5 L發酵罐分批補料發酵曲線Fig. 8 Fed-batch fermentation profiles of the engineered strain HYP15 in a 5-L bioreactor
3 結 論
本研究構建了1 株無質粒、從頭高效合成反式-4-羥基-L-脯氨酸的大腸桿菌基因工程菌。工程菌的構建過程包括:1)通過引入木糖啟動子控制的T7 RNA聚合酶基因,在大腸桿菌W3110中建立木糖誘導的基因表達系統;2)通過刪除L-脯氨酸脫氫酶基因putA,阻斷L-脯氨酸的降解途徑;3)通過L-脯氨酸操縱子和吡啶核苷酸轉氫酶基因整合,增強了L-脯氨酸的合成代謝;4)篩選到高活性的L-脯氨酸-4-羥基化酶基因p4H1;5)通過L-脯氨酸-4-羥基化酶基因整合,強化反式-4-羥基-L-脯氨酸的合成代謝。以葡萄糖為碳源進行分批補料發酵40 h,工程菌HYP15的反式-4-羥基-L-脯氨酸產量達到48.6 g/L,糖酸轉化率和生產強度分別達到了21.6%和1.22 g/(Lg h)。
與現有報道的反式-4-羥基-L-脯氨酸生產菌相比,該工程菌,具有以下優勢:1)菌株基因型明確,遺傳穩定性好;2)不含質粒表達載體,避免抗生的使用;3)發酵周期短,生產強度高。工程菌具有良好的工業化前景,為反式-4-羥基-L-脯氨酸發酵法生產提供了理論支持。
猜你喜歡
礦山安全信息(2022年40期)2022-04-07 02:16:52
當代水產(2021年10期)2021-12-05 16:31:48
今日農業(2021年14期)2021-11-25 23:57:29
今日農業(2021年13期)2021-08-14 01:37:56
石油與天然氣地質(2021年1期)2021-02-22 14:14:44
今日農業(2020年20期)2020-11-26 06:09:10
中國果業信息(2019年10期)2019-11-13 01:21:34
中國化肥信息(2019年2期)2019-01-18 15:24:35
中國化肥信息(2019年1期)2019-01-17 21:31:12
中國化肥信息(2019年4期)2019-01-17 18:47:06
