金屬Sr、Fe 摻雜對LaCoO3 催化氧化碳煙及抗硫性能影響
2020-02-11 09:01:34吳艾純喬智威李樹華
無機化學學報 2020年1期
魏 煒 吳艾純 喬智威 李樹華 梁 紅 彭 峰
(廣州大學化學化工學院,能源與催化研究所,廣州 510006)
0 引 言
柴油機以其良好的燃料利用率、 耐久性和CO2排量低等優點,廣泛應用于輪船、汽車、貨車、發電廠等大型設備[1]。 然而,由于油品燃燒不完全導致柴油機尾氣中含有大量碳煙(soot)顆粒,對環境和人體健康造成較大危害[2-3]。 隨著國家對移動源尾氣排放法規的日趨嚴格, 僅機內凈化技術已難以達到當前的排放標準,因此,開發一種性能優良的柴油機尾氣凈化技術顯得勢在必行。 目前針對尾氣中碳煙去除主要安裝堇青石DPF(顆粒過濾器)過濾[4],但隨著碳煙在DPF 壁內積累易導致發動機排氣背壓增加,使柴油機運行工況惡化并產生更多污染物, 降低其運行壽命,而碳煙在發動機(200~400 ℃)中極難自身氧化分解, 因此針對這一難題研究一種催化氧化碳煙的高效催化劑顯得勢在必行。另一方面,由于當前原油煉制中脫硫的不完全性導致柴油機尾氣中含有一定SO2,易使催化劑中毒失活,且有關催化劑抗硫性能和中毒機理亦是當前環境催化領域一大熱點。因此,研究催化氧化碳煙性能和抗硫中毒機理對設計、指導高效催化劑開發具有較大的理論和實際意義。
目前在各類移動源尾氣催化劑中, 主要有金屬氧化物[5]、貴金屬[6]、尖晶石[7]和鈣鈦礦型[8]催化劑。 其中鈣鈦礦催化劑相較其它催化劑具有價格便宜、熱穩定性好,催化活性高等優點,被廣泛應用于環境催化領域[9-10]。 鈣鈦礦命名來源于稀土金屬氧化物CaTiO3,其分子式為ABO3[11];通過金屬離子摻雜可使催化劑性能得到較好調變,通常鈣鈦礦A 位可被堿金屬或堿土金屬取代,B 位可被過渡金屬取代而保持其晶體結構不變;其中,堿土金屬摻雜對鈣鈦礦催化劑性能調變至關重要。Dai 等[12]通過模板法制備Sr摻雜的Eu1-xSrxFeO3催化劑, 研究表明Sr 摻雜能增加催化劑表面吸附氧濃度和低溫還原性能;Liu 等[13]采用共沉淀法制備La1-xSrxMnO3, 通過SEM 表征發現Sr 摻雜能形成超細納米顆粒,對催化氧化CH4表現出較高催化活性;Gao 等[14]制備納米鈣鈦礦催化劑La1-xSrxTiO3, 并用于催化氧化碳煙具有較好的催化活性,表明金屬Sr 摻雜鈣鈦礦催化劑具有較好的性能調變潛力。 另一方面,Fe 基氧化物催化劑在環境催化領域也有廣泛應用,如FeTiOx[15]、Fe2O3-SiO2[16]、WOx/Fe2O3[17];Li 等[18]利 用 檸 檬 酸 配 位 法 制 備 了La0.9K0.1Co1-xFexO3,研究表明Fe 摻雜能提高晶格氧活性,極大促進對碳煙的催化氧化,這表明Fe 調變催化劑性能有較好應用前景。 催化劑抗硫性能也是工業尾氣催化劑的重要指標, 研究表明SO2易使催化劑中毒失活, 且有關抗硫機理也是當前熱點。 Jin等[19]制備了Mn/TiO2和Mn-Ce/TiO2催化劑,研究表明當SO2引入Mn/TiO2時其NO 轉化率從90%降低至30%;Yu 等[20]制備了PrOx(0.2)-MnOx/SAPO-34 催化劑, 研究表明當SO2氣氛出現時NO 轉化率降低81.0%;Li 等[21]制備了CeO2-MnOx催化劑,研究表明當通入SO2時NO 轉化率降低至75%, 即使停止通入SO2時其催化活性也未能恢復, 表明SO2對催化劑活性有較大影響,且較難活化恢復。 Liu 等[22]利用Sm 摻雜CeO2-TiO2催化劑,通過XPS 和DFT (密度泛函理論) 計算表明,SmCeTi 催化劑抗硫性來源于Sm2++Ce4+?Sm3++Ce3+氧化還原反應,有效地抑制了電子從SO2向Ce4+遷移, 使催化劑具有較好的抗硫能力;Jiang 等[23]制備了V2O5共摻雜Mn-Ce(0.4)-V/AC 催化劑, 研究表明催化劑抗硫性來源于V2O5摻雜形成的酸性和Mn-Ce 固溶體的分散,有效抑制了SO2對Mn-Ce 的硫酸化;Yu 等[24]制備的MxCe1-xOδ/3DOM-mSiO2催化劑表現出良好的催化燃燒碳煙活性,通過XRD、Raman、DFT 等證明,SO2中毒主要由于催化劑吸附SO2與活性氧及金屬原子反應形成硫酸鹽物種,導致催化活性降低,這表明SO2對催化劑活性有較大影響, 且有關催化劑抗硫性能優化和機理也未十分清晰, 這也是當前催化劑抗硫研究一大熱點。因此,針對催化劑性能優化及抗硫機理研究對未來移動源尾氣凈化具有較好借鑒意義。
本工作采用檸檬酸-EDTA 配位法制備4 種性能較佳的鈣鈦礦催化劑La1-xSrxCo1-yFeyO3(x=0,0.1;y=0,0.3),分別研究金屬Sr、Fe 摻雜對催化劑物理化學性能調變和抗硫中毒機理, 以期對未來機動車尾氣催化劑的設計、開發提供有效理論支撐。
1 實驗部分
1.1 催化劑的制備
通過檸檬酸-EDTA 配位法制備鈣鈦礦催化劑La1-xSrxCo1-yFeyO3, 分別記為LaCoO3、La0.9Sr0.1CoO3(LaSrCoO3)、LaCo0.7Fe0.3O3(LaCoFeO3)、La0.9Sr0.1Co0.7Fe0.3O3(LaSrCoFeO3)。 即按化學計量比稱取La(NO3)3·6H2O、Sr(NO3)2、Co(NO3)2·6H2O、Fe(NO3)3·9H2O、檸 檬 酸 和EDTA,保持總金屬離子、檸檬酸、EDTA 的物質的量之比為1∶1∶0.2,并用20 mL 去離子水溶解使上述金屬硝酸鹽充分混合。將混合溶液置于80 ℃恒溫水浴鍋中加熱攪拌,至溶液逐漸形成粘稠狀凝膠;后轉移至150 ℃恒溫油浴中保溫2 h 使凝膠充分膨脹得到疏松多孔的干凝膠, 再轉移至120 ℃恒溫干燥箱中干燥過夜得催化劑前驅體。經破碎、研磨轉移至真空管式爐中, 在空氣氣氛下以5 ℃·min-1升溫至400℃焙燒2 h,再升溫至750 ℃焙燒5 h,得黑色催化劑粉體。將上述催化劑于300 ℃下在以N2為平衡氣的0.08%(V/V)SO2中硫化處理2 h, 空速 (GHSV)為10 000 h-1[25]。 硫化后的催化劑分別記為LaCoO3-S、LaSrCoO3-S、LaCoFeO3-S、LaSrCoFeO3-S。 使用后的催化劑分別記為LaCoO3-U、LaSrCoO3-U、LaCoFeO3-U、LaSrCoFeO3-U。
1.2 催化劑的表征
1.2.1 XRD 分析
催化劑的XRD 分析使用荷蘭PANalytical 公司生產的PW3040/60 型X 射線衍射儀。 采用Co Kα(λ=0.179 03 nm) 輻射源, 管電壓40 kV, 管流40 mA,衍射角2θ=10°~90°,掃描步長0.02°。
1.2.2 FT-IR 表征
傅利葉紅外光譜測試(FT-IR)在Thermo Nicolet公司生產的Nexus FT-IR 紅外光譜儀上進行。 光譜分辨率4 cm-1,掃描范圍為4 000~400 cm-1。 催化劑樣品與KBr 按質量比1∶100 混合并充分干燥研磨并壓片測試。
1.2.3 SEM 表征
采用日本JEOR 公司生產的JSM-7001F 型熱場發射掃描電子顯微鏡對催化劑表面形貌進行SEM分析。測試前對樣品進行噴金處理以增強其導電性,掃描電鏡工作電壓為10 kV。 采用能量分散譜儀(EDS)分析樣品的元素組成。
1.2.4 程序升溫還原(H2-TPR)表征
程序升溫還原使用天津先權公司TP 5080 型催化劑評價裝置,50 mg 催化劑在N2氣氛下從室溫以10 ℃·min-1程序升溫至400 ℃保持1 h,后降至室溫混氫30 min 待穩定基線, 氣氛為95% N2和5% H2(V/V),升溫速率為30 ℃·min-1。 程序升溫至900 ℃,使用TCD 檢測器記錄,水氣使用干燥分子篩吸附。
1.2.5 XPS 表征
催化劑的元素價態掃描在Thermo ESCALAB 250Xi 型X 射線光電子能譜儀(XPS)上進行,采用Al Kα(hν=1 486.6 eV)作為光源,真空度為5×10-8Pa,以污染碳(C1s:284.8 eV)用于校準作為能量標準,結合能誤差為±0.2 eV。 采用Gaussian-Lorentzian 法及Shirley 扣除背景,計算表面原子含量和價態分布。
1.2.6 程序升溫脫附(SO2-TPD)表征
程序升溫脫附使用天津先權公司TP 5080 型催化劑評價裝置,取150 mg 催化劑在He 氣氛下從室溫以10 ℃·min-1程序升溫至400 ℃保持1 h, 后降至室溫切換3%(V/V)SO2氣氛吸附30 min,再切換為He 氣充分吹掃表面物理吸附的SO2至基線穩定。在30 mL·min-1純He 氣氛中程序升溫至900 ℃, 使用TCD 檢測器記錄信號。
1.2.7 熱重表征
實驗使用PerkinElmer 公司生產的TGA 4000型熱重分析儀,取30 mg 催化劑在N2氣氛下從室溫以10 ℃·min-1程序升溫至800 ℃。
1.3 催化劑活性評價
采用天津先權公司生產的連續氣固相反應裝置(WFS-3015)評價催化氧化碳煙活性。 采用緊密接觸以克服反應中不均勻傳熱和傳質, 按1∶9 質量比稱取碳煙和催化劑, 充分研磨并攪拌, 置入干燥箱干燥, 以保持各催化劑具有相同接觸條件, 碳煙選用Degussa 公司生產的Printex-U 碳代替柴油機尾氣中碳煙顆粒。 反應使用180 mg 催化劑,氣氛為0.15%(V/V)NO+5%(V/V)O2+N2平衡氣, 混合氣總流量為100 mL·min-1,反應空速(GHSV)為15 000 h-1,在溫度為200~500 ℃內評價其催化氧化碳煙活性。 碳煙氧化產生的CO2及氣氛中O2的濃度采用安捷倫科技有限公司生產的GC 7820A 型氣相色譜檢測。 本工作中, 碳煙開始燃燒溫度定義為起燃溫度(Ti),碳煙最大燃燒速率時溫度定義為(Tm),碳煙燃盡溫度定義為(Tf)。式(1)為碳煙燃燒產生的CO2在氣氛中所占的濃度(CCO2),該濃度越大表明催化燃燒碳煙速率越快,一般用于評價催化氧化碳煙活性;其Ti、Tm和Tf越低,表明催化劑活性越高。

其中,αCO2為反應器出口氣體中的CO2濃度;αO2為反應器出口氣體中的O2濃度。
2 結果與討論
2.1 催化氧化碳煙活性與抗硫性能
圖1 為催化劑催化氧化碳煙活性曲線, 碳煙燃燒在無催化劑時Tm為586 ℃, 在柴油機尾氣中(200~400 ℃) 極難自身氧化分解。 當反應引入LaCoO3催化劑時, 碳煙催化氧化溫度降低33.8%,其Tm為387 ℃; 當進一步摻雜金屬Sr 后, 發現LaSrCoO3具有較高的催化燃燒碳煙活性(碳煙氧化溫度降低40.6%),其Tm僅為347 ℃,表明Sr 摻雜有利于提高對碳煙催化活性。 當催化劑同時摻雜Sr、Fe 時,催化氧化碳煙活性并未進一步提高(Tm為368℃),表明Sr、Fe 摻雜抑制了催化氧化碳煙活性提高。另一方面,催化劑抗硫性能也是重要評價指標。 表1表明催化劑經過SO2硫化后,SO2對LaCoO3活性有較大影響(Tm升高24.5%), 其Tm為482 ℃; 而當LaCoO3同時摻雜Sr、Fe 后, 其催化氧化碳煙活性(Ti=320 ℃,Tm=361 ℃,Tf=460 ℃)變化較小,表明同時摻雜Sr、Fe 有利于改善LaCoO3抗硫中毒能力。綜上所述,采用檸檬酸-EDTA 配位法制備的催化劑具有較好的催化氧化碳煙活性,其中Sr 摻雜有較好的催化碳煙活性; 同時摻雜Sr、Fe 雖未使活性進一步提高,但有利于提高其抗硫性能。
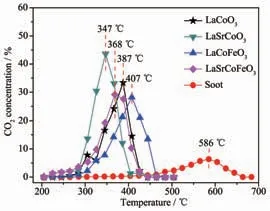
圖1 催化氧化碳煙活性曲線Fig.1 Catalytic oxidation activity curve of soot
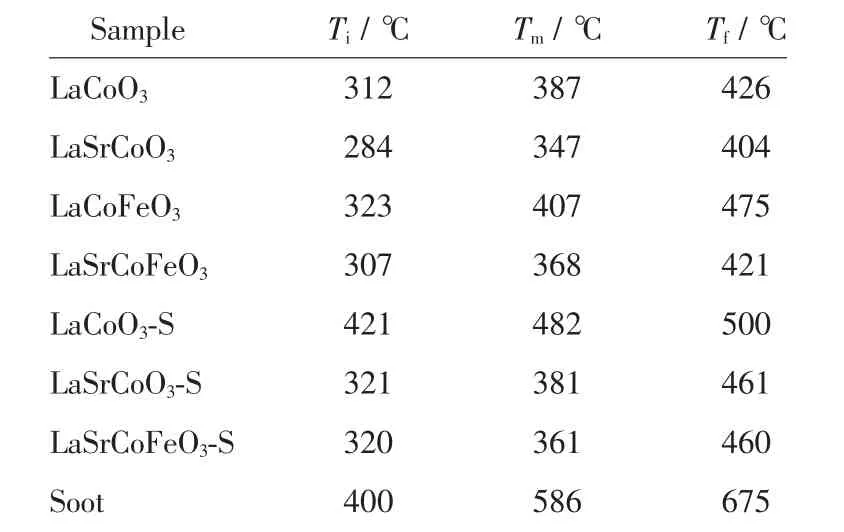
表1 催化劑硫化前后催化氧化碳煙活性對比Table 1 Comparison of catalytic oxidation soot activity before and after catalyst vulcanization
2.2 XRD 分析
為研究金屬摻雜和SO2對催化劑結構影響,對硫化前后的La1-xSrxCo1-yFeyO3催化劑進行XRD 表征。 圖2(A)表明各催化劑主要特征衍射峰與LaCoO3(PDF No.75-0279)標準卡一致,表明LaCoO3催化劑摻雜Sr、Fe 后仍保持完整鈣鈦礦結構。 值得注意的是,La1-xSrxCo1-yFeyO3-S 的特征峰與新鮮催化劑基本一致,無明顯硫酸鹽衍射峰出現,表明硫化后對鈣鈦礦晶型無明顯破壞。 由圖2(B)可知,催化劑摻雜Sr、Fe 后,衍射峰向低角度偏移。根據Bragg′s Law,由于Sr2+(0.118 nm)、Fe3+(0.065 4 nm) 離子半徑大于La3+(0.103 2 nm)、Co3+(0.061 nm),使Sr、Fe 能成功進入鈣鈦礦晶格結構。而相較新鮮催化劑,硫化后衍射峰進一步向低角度偏移, 這可能由于SO2與摻雜金屬或表面吸附氧結合而引起的晶格膨脹; 當催化劑同時摻雜Sr、Fe 時,La1-xSrxCo1-yFeyO3-S 衍射峰向高角度偏移, 這可能由于SO2與摻雜金屬結合導致的晶格收縮。 圖2(C)為使用前后催化劑XRD 圖對比,使用前后催化劑晶型結構無明顯變化, 表明各催化劑具有較好結構穩定性。
2.3 FT-IR 分析
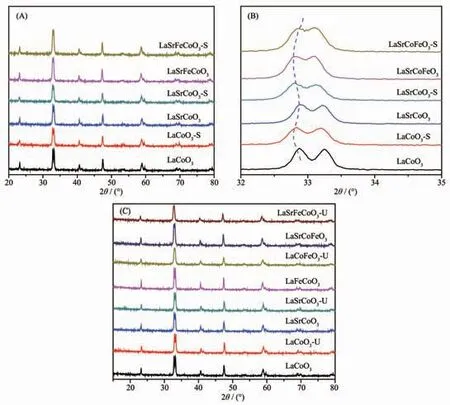
圖2 La1-xSrxCo1-yFeyO3 催化劑的XRD 圖Fig.2 XRD patterns of La1-xSrxCo1-yFeyO3 catalysts
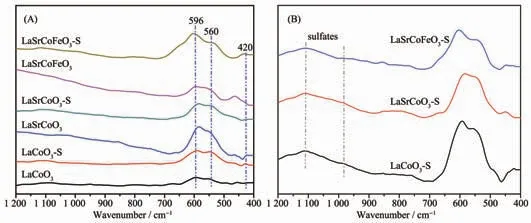
圖3 La1-xSrxCo1-yFeyO3 催化劑的FT-IR 譜圖Fig.3 FT-IR spectra of La1-xSrxCo1-yFeyO3 catalysts
為進一步研究金屬摻雜對La1-xSrxCo1-yFeyO3催化劑的結構影響, 對各催化劑的FT-IR 圖譜進行研究(圖3)。 鈣鈦礦ABO3基本結構為BO6八面體,八面體之間的間隙原子是A 位陽離子,BO6紅外譜圖有6 種振動模式。 如果3 對B-O 鍵具有相同鍵長,即BO6八面體具有高對稱性時,伸縮振動(ν3)是非紅外活性的;當BO6八面體對稱性較低時,B-O 鍵伸縮振動(ν3)則具有紅外活性[26]。 由圖可知,所有樣品主要存在3 種紅外振動, 分別在596、560 和420 cm-1處。 研究表明波數在596 和560 cm-1處振動峰歸屬于CoO6八面體中的2 種Co-O 伸縮振動, 而420 cm-1處的振動歸屬于CoO6中Co-O 鍵的彎曲振動[9];該結果進一步證明制備的La1-xSrxCo1-yFeyO3催化劑樣品具有明顯鈣鈦礦型氧化物(ABO3)結構。 此外,由圖3(B)可知,催化劑硫化后在975、1 074、1 137 cm-1處形成明顯混合峰,這主要歸屬為硫酸鹽物種(SO32-和SO42-)在催化劑表面的沉積[27]。綜上所述,Sr、Fe 摻雜仍能保持完整鈣鈦礦結構, 且SO2對鈣鈦礦結構無明顯影響,SO2主要以硫酸鹽形式存在。
2.3 結構分析
為研究金屬摻雜及SO2對催化劑微觀形貌影響, 對La1-xSrxCo1-yFeyO3催化劑進行SEM 表征。LaCoO3顆粒形狀大小不均且出現一定程度團聚;當樣品摻雜Sr 后,其顆粒分散度較好,無明顯燒結現象,且顆粒呈球形分布,尺寸大小在110 nm 左右,其催化劑顆粒尺寸與真實碳煙顆粒(70~100 nm)處于同一量級[28],這使得兩者可達到較大接觸面積,為反應提供必要氣-固-固接觸位點。 而同時摻雜Sr、Fe 時,顆粒尺寸有一定變大趨勢,這可能是導致催化氧化碳煙活性下降原因之一。 當催化劑硫化后,LaCoO3-S 顆粒進一步團聚在一起,且顆粒之間呈無規則堆疊,這不利于催化劑與氣相接觸,而LaSrCoO3-S、LaSrCoFeO3-S 與新鮮催化劑相比, 顆粒分布和尺寸無明顯變化, 表明SO2對催化劑表面顆粒狀態及形貌沒有明顯破壞, 故Sr、Fe 摻雜提高了催化劑對SO2的耐受性。圖5 為催化劑硫化后的EDS-mapping照片,發現硫化后催化劑LaSrCoFeO3-S 中S 原子百分含量少于LaCoO3催化劑(XPS 將進一步確定S 的相對含量),表明Sr、Fe 摻雜有利于抑制SO2在催化劑表面沉積, 降低SO2對催化劑毒化; 其它各元素(La、Co、O、Sr、Fe)在催化劑中亦有較好分布,表明元素間可能存在一定相互作用, 有利于催化活性提高和抗硫性能改善。

圖4 La1-xSrxCo1-yFeyO3 催化劑的SEM 圖Fig.4 SEM images of La1-xSrxCo1-yFeyO3 catalysts

圖5 La1-xSrxCo1-yFeyO3 催化劑硫化后的EDS-mapping 照片Fig.5 EDS-mapping photographs of La1-xSrxCo1-yFeyO3 catalysts after vulcanization
2.4 H2-TPR 表征
為研究金屬摻雜和SO2對催化劑氧化還原性能影響, 測定了La1-xSrxCo1-yFeyO3催化劑H2-TPR 譜圖(圖6)。 研究表明H2不僅能還原催化劑中變價金屬離子,其表面氧物種亦能參與還原反應,這在一定程度上可反映催化劑中表面氧物種活性[29]。 LaCoO3有2 個主要的還原峰, 在440 ℃處還原峰歸屬為Co3+還原為Co2+和表面吸附氧物種,622 ℃處還原峰歸屬為Co2+還原為Co 和晶格氧物種[1]。 當Sr 摻雜LaCoO3后還原峰向低溫偏移, 表明Sr 摻雜有利于提高催化劑低溫還原性能和表面氧物種活性。 當催化劑同時摻雜Sr、Fe 時,其高溫還原峰消失,低溫還原峰并向左偏移,表明Sr、Fe 存在較強的相互作用,這導致催化劑中價態環境變化, 有利于增加低溫還原性能和表面氧物種活性。 La1-xSrxCo1-yFeyO3硫化后的還原性能有一定程度的降低,其中硫化對LaCoO3還原性能影響較大(低溫還原峰溫升高130 ℃),表明SO2可能減少催化劑中Co2+/Co3+及表面氧物種含量;而LaSrCoFeO3-S 低溫還原性能雖有一定降低(低溫還原峰溫為455 ℃), 但低溫還原性能仍優于LaSrCoO3(低溫還原峰溫500 ℃)和LaCoO3(低溫還原峰溫為556 ℃),表明Sr、Fe 同時摻雜對低溫還原性能和抗硫性能有較大改善。綜上所述,催化劑低溫還原性能和抗硫能力順序為:LaSrCoFeO3>LaSrCoO3>LaCoO3, 催化劑中毒可能主要來源于SO2對Co2+/Co3+和表面吸附氧的硫酸化。
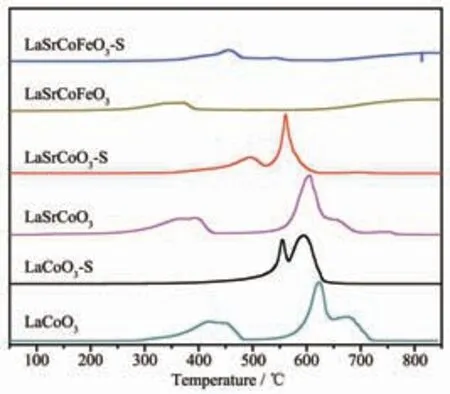
圖6 催化劑La1-xSrxCo1-yFeyO3 的H2-TPR 譜圖Fig.6 H2-TPR profiles of La1-xSrxCo1-yFeyO3 catalysts
2.5 XPS 分析
為進一步研究催化劑中硫物種價態及各元素化學環境,研究了S2p 的XPS 價態分布(圖7(A))。 研究表明,168.4 和169.6 eV 處的譜峰分別歸屬為S6+的2p3/2和2p1/2;在~168.9 和170.1 eV 處分別歸屬為S4+的2p3/2和2p1/2[30-31]。 當La1-xSrxCo1-yFeyO3硫化后,S2p的分峰擬合圖表明催化劑中SO2主要以SO32-和SO42-形式存在, 這也與FT-IR 結果一致。 表2 表明LaSrCoFeO3-S 表面硫的原子百分含量(3.18%) 少于LaSrCoO3(4.00%)和LaCoO3(4.06%),表明同時摻雜Sr、Fe 有利于抑制SO2在催化劑表面沉積, 這也與EDS-mapping 結果一致。 綜上所述,同時摻雜Sr、Fe有利于減少SO2在催化劑表面的沉積, 且SO2在催化劑上以硫酸鹽(SO32-和SO42-)形式存在。
圖7(B)為La1-xSrxCo1-yFeyO3催化劑硫化前后O1s的XPS 譜圖, 其結合能及表面吸附氧含量列于表3中,研究表明,低結合能(~529.5 eV)處的O1s 峰歸屬為晶格氧物種Oβ(O2-),高結合能(~532.0 eV)處歸屬于表面吸附氧物種Oα(O2-和O-)。 Oα和Oβ通常存在于缺陷型氧化物中[32-33],且表面吸附氧相對晶格氧具有較好的活性和移動性, 這對碳煙催化氧化至關重要。 當LaCoO3摻雜Sr 后形成較多表面吸附氧(58.30%), 表明Sr 摻雜有利于形成豐富的表面吸附氧和氧空位,有利于提高催化氧化碳煙活性,這也與圖1 的活性結果一致。 當Sr、Fe 同時摻雜時其表面吸附氧含量(60.60%)增加,結合H2-TPR 結果可知,這主要是因為金屬摻雜導致的電荷轉移使表面吸附氧含量增加。 值得注意的是,當各催化劑硫化后,表面吸附氧的含量增加, 這主要由于SO2與催化劑中金屬離子及表面吸附氧反應形成硫酸鹽物種導致[23]。當LaCoO3硫化后,會形成較多的硫酸鹽物種(S元素的原子百分含量90.29%),表明LaCoO3硫化后對表面吸附氧有較大消耗; 而同時摻雜Sr、Fe 時表面吸附氧含量相對于新鮮催化劑變化較小,表明Fe摻雜對表面吸附氧有一定保護, 這也與EDSmapping 和表3 結果一致。 綜上所述,催化劑中毒可能主要來源于SO2對催化劑中Co2+/Co3+及表面吸附氧的硫酸化。
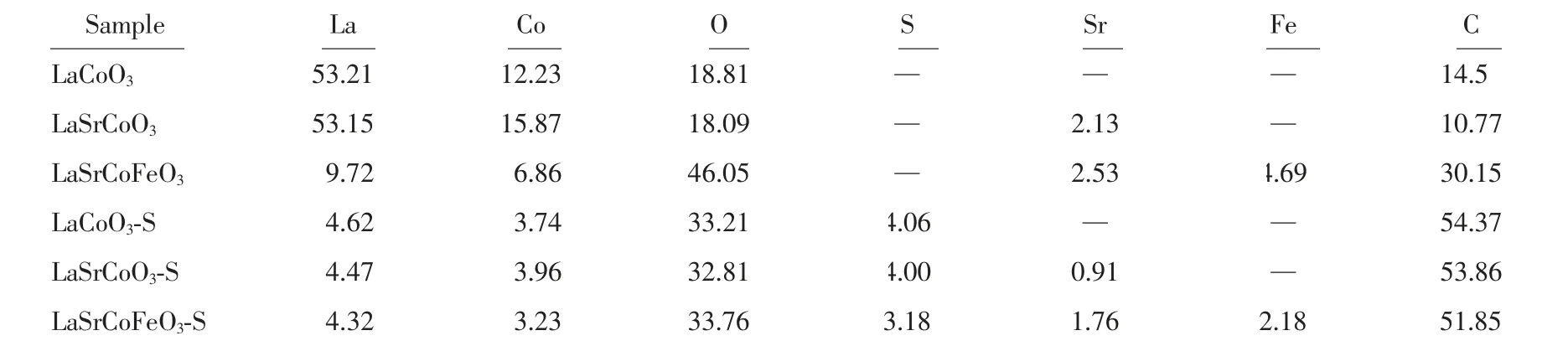
表2 La1-xSrxCo1-yFeyO3 催化劑硫化前后各元素相對原子百分含量Table 2 Relative atomic contents of elements for catalyst La1-xSrxCo1-yFeyO3 after vulcanization%
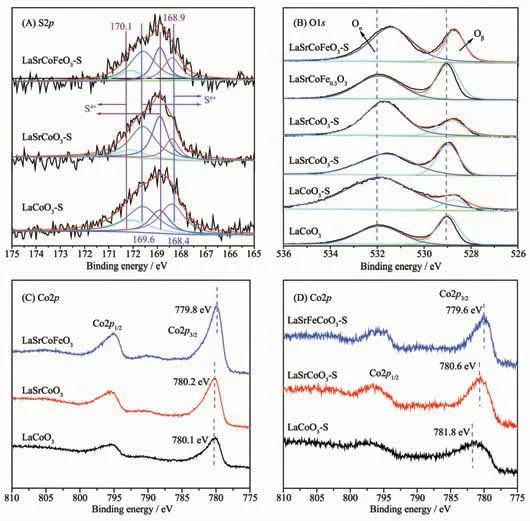
圖7 La1-xSrxCo1-yFeyO3 硫化前后的XPS 譜圖Fig.7 XPS spectra of La1-xSrxCo1-yFeyO3 catalysts
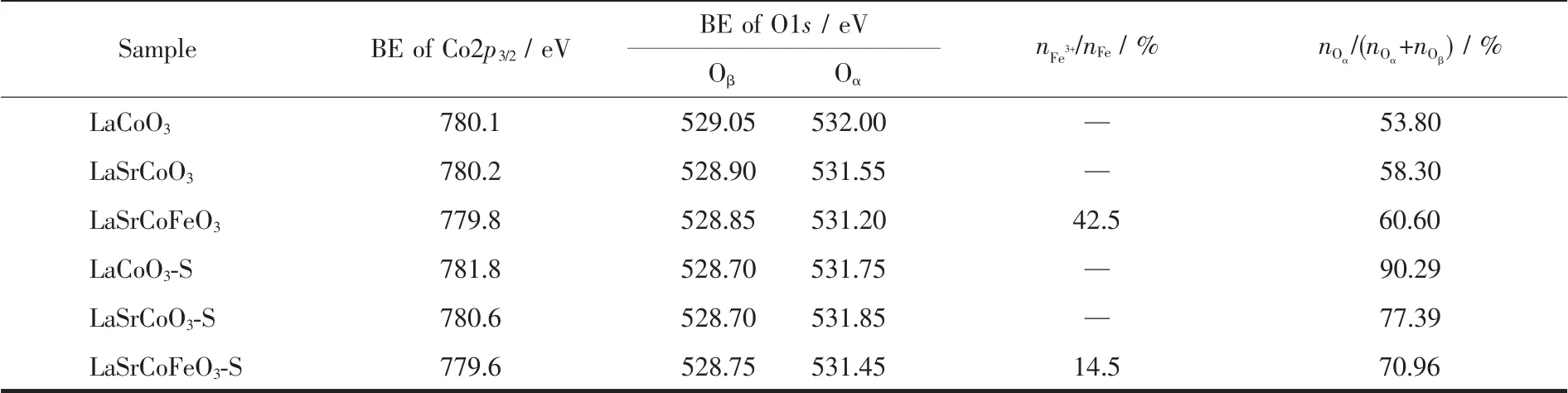
表3 La1-xSrxCo1-yFeyO3 催化劑硫化前后Co2p3/2, O1s 結合能和Fe3+、Oα 百分含量Table 3 Binding energies (BE) of Co2p3/2 and O1s, and the content of Fe3+and Oα for the La1-xSrxCo1-yFeyO3 catalysts
圖7(C,D)為La1-xSrxCo1-yFeyO3催化劑硫化前后Co2p 的XPS 譜圖。 各催化劑均明顯表現出2 個Co2p 肩峰,分別歸屬為Co2p1/2和Co2p3/2。 LaCoO3催化劑Co2p3/2的結合能為780.10 eV,而同時摻雜Sr、Fe 后,Co2p3/2向低結合能移動至779.85 eV,這是由于Fe 摻雜改變了鈣鈦礦的電荷平衡,而為補償鈣鈦礦氧化物使其電中性, 催化劑會形成更多高價Fe4+和氧空位以平衡電荷,這有利于催化氧化碳煙反應。值得注意的是,LaCoO3和LaSrCoO3硫化后,Co2p3/2向高結合能偏移,表明SO2對催化劑Co2p 價態環境有一定影響, 結合H2-TPR 可知, 這是由于SO2對Co2+/Co3+及表面吸附氧的硫酸化,導致催化劑結合能偏移。進一步由表3 可知,LaSrCoFeO3-S(14.5%)相對新鮮的LaSrCoFeO3(42.5%) 中的Fe3+含量下降65.8%,表明LaSrCoFeO3-S 抗硫性可能由于Fe3+與表面吸附氧及SO2共同作用形成了Fe2(SO3)3、Fe2(SO4)3。另一方面,LaSrCoFeO3-S 中Co2p3/2結合能也相對新鮮催化劑LaSrCoFeO3變化較小, 表明Fe 摻雜有利于對Co2p 價態環境的保護,即同時摻雜Sr、Fe 形成的Fe3+有助于減少SO2對活性位點表面吸附氧和Fe4+毒化。
2.5 SO2-TPD 和熱重-微商熱重分析
為進一步研究金屬摻雜對吸脫附SO2的影響及硫酸鹽的形式, 對La1-xSrxCo1-yFeyO3催化劑的SO2-TPD 曲線進行研究(圖8(A))。 在50~200 ℃為SO2物理吸附態,250~500 ℃為中度硫酸鹽物種分解,大于650 ℃為強粘結硫酸鹽物種的分解[34-35]。LaCoO3催化劑中的SO2在整個溫度范圍內主要呈物理及中度吸附, 即SO2與表面吸附氧形成弱或中度硫酸鹽覆蓋催化劑活性位點,導致催化劑失活,這也和XPS 結果一致。當催化劑同時摻雜Sr、Fe 時,在低溫范圍內對SO2吸附減少, 有利于降低SO2對表面吸附氧的毒化, 并在高溫形成2 個脫附峰, 即670~800 ℃和800~860 ℃,這主要歸屬為SO2與Fe3+及表面吸附氧反應形成的強粘結硫酸鹽Fe2(SO3)3和Fe2(SO4)3。圖8(B)為LaSrCoFeO3-S 催化劑的熱重-微商熱重分析(TG-DTG) 曲線, 其在480 ℃處出現一個DTG 失重峰,這主要歸屬為Fe2(SO3)3和Fe2(SO4)3的分解[36],表明LaSrCoFeO3催化劑抗硫性主要來源于形成的Fe2(SO3)3和Fe2(SO4)3保護了活性位點的表面吸附氧和Fe4+。
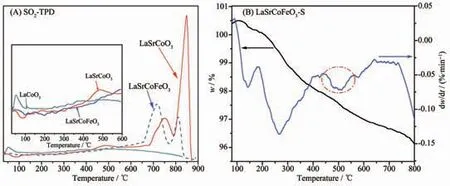
圖8 La1-xSrxCo1-yFeyO3 催化劑的(A) SO2-TPD 和(B) TG-DTGFig.8 (A) SO2-TPD and (B) TG-DTG profiles of La1-xSrxCo1-yFeyO3 catalysts
3 結 論
本研究利用檸檬酸-EDTA 配位法制備了一系列鈣鈦礦催化劑La1-xSrxCo1-yFeyO3, 展現出了較好的催化氧化碳煙活性。 Sr 摻雜LaCoO3有利于形成更多的表面吸附氧(O2-、O-)和氧空位,改善了低溫氧化還原性能,提高了低溫催化氧化碳煙活性,其Ti和Tm僅為284 和347 ℃。 摻雜Sr、Fe 后低溫氧化還原性能進一步提高,并形成更多的Fe4+離子,這有利于改善催化氧化碳煙活性。 催化劑SO2中毒主要由于Co2+/Co3+和表面吸附氧的硫酸化(SO32-、SO42-);同時摻雜Sr、Fe 后兩者之間形成的相互作用能有效抑制硫酸鹽在催化劑表面的沉積, 減少SO2對催化劑的毒化。 通過XPS 和SO2-TPD 發現,催化劑抗硫性主要來源于金屬離子Fe3+與SO2結合形成硫酸鹽(Fe2(SO3)3和Fe2(SO4)3),使SO2減少對活性組分表面吸附氧和Fe4+的毒化。 通過TPO 發現硫化后的LaSrCoFeO3仍具有較好的催化氧化碳煙活性, 其Ti和Tm分別為320 和361 ℃,表明Sr、Fe 同時摻雜具有較好的低溫催化氧化碳煙活性和良好的抗硫性能。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
