基于財務視角分析中國飛鶴的發展
2022-10-03 06:09:56吳國蕊
中國乳業 2022年9期
吳國蕊
南平建設集團信達供應鏈有限公司,福建建陽 354200
0 引言
2008年的“三鹿牌嬰幼兒奶粉”事件重創了我國消費者對乳制品的消費信心,但經過13年的發展,消費者、乳品企業、監管層面對于中國嬰幼兒配方奶粉(簡稱“嬰配粉”)的監管力度愈發嚴格,根據國家統計局數據顯示,2020年和2021年嬰配粉抽檢合格率分別為99.89%和99.88%。中國飛鶴有限公司(簡稱“中國飛鶴”)也在13 年中獲得了發展的好機會,成為中國奶粉行業的龍頭企業。
中國飛鶴以“更適合中國寶寶體質”為定位,以“五十多年安全零事故”為依托,為產品做出了安全承諾,為重構中國消費者心智邁出了第一步。2019年11月中國飛鶴在港交所成功上市,成為中國歷史上首發市值最大的奶粉企業,榮膺國產嬰配粉的第一品牌。
中國飛鶴是中國優秀奶粉企業的典型代表。通過分析中國飛鶴的財務狀況,能夠更加直觀地體現企業的發展狀況和存在問題。因此本文基于財務視角分析數據,為中國飛鶴提供一些合理的建議,也通過分析中國飛鶴的發展,為其他中國奶粉企業提供借鑒和啟發。
1 中國飛鶴主要財務情況分析
1.1 中國飛鶴的資產負債和利潤分析
中國飛鶴2016—2020年的資產負債表和利潤表分別見表1和表2。中國飛鶴的資產從2016年的46.95億元到2020年的283.23 億元,翻了6 倍,企業的規模不斷壯大;負債從23.18 億元到2020年的91.37 億元,翻了3.94 倍,負債增長速度小于資產增長速度,負債增長后,負債的利息能夠從稅前利潤扣除,減少企業所得稅應納稅所得額,從而減少了企業應納所得稅,企業的負債額越大,負債利息抵稅效應越大,越有利于企業的進一步發展;股東權益從23.77 億元到2020年的191.86 億元,翻了8 倍,企業股東投入加大,經營獲利也不斷增加,企業發展加快;營業收入從37.24 億元到185.92 億元,翻了4.99 倍,商品銷量不斷增長,業務不斷擴大;凈利潤從4.06 億元到74.37 億元,翻了18 倍,凈利潤增長速度大于營業收入增長,企業收益顯著增加。以上數據表明,中國飛鶴2016—2020年企業運營良好,經營成果突出,企業在不斷地發展壯大。

表1 利潤表主要數據 單位:億元

表2 資產負債表主要數據 單位:億元
1.2 中國飛鶴償債能力分析
償債能力是指企業用資產來清償企業債務的能力,主要分為償還長期債務與短期債務的能力[1]。中國飛鶴償債能力指標如表3所示。
從表3可以看出,2016—2020年中國飛鶴的流動比率逐年增長。2016—2018年在2以下;2019年和2020年在2以上。在通常情況下,企業的流動比率在2比較適合,說明中國飛鶴的短期償債能力不斷增強。

表3 中國飛鶴償債能力指標
2016—2020年中國飛鶴的速動比率均超過了1;2019年和2020年中國飛鶴的速動比率分別達到2.23、2.43,與2019年和2020年的流動比率較為接近。在通常情況下,企業的流動比率在1比較適合,說明中國飛鶴的變現能力在不斷變強,現金持有在增多,可以進一步提高企業資金的利用效率。
2016—2019年中國飛鶴的資產負債率在正常范圍40%~60%內;2020年中國飛鶴的資產負債率為32%,低于正常范圍,說明可能是公司融資管理相對保守,財務杠桿作用沒能充分發揮[2]。
2016—2020年中國飛鶴的產權比率均小于1.2,其中2020年產權比率為0.48。在通常情況下,企業的產權比率的標準值是1.2,說明中國飛鶴的長期償債能力強,債權人投資風險低,受到保障的程度高。
1.3 中國飛鶴的營運能力分析
營運能力是指企業利用資產賺取利潤的能力。企業營運能力可以通過應收賬款周轉率、存貨周轉天數、存貨周轉次數來分析[1]。中國飛鶴營運能力指標如表4所示。
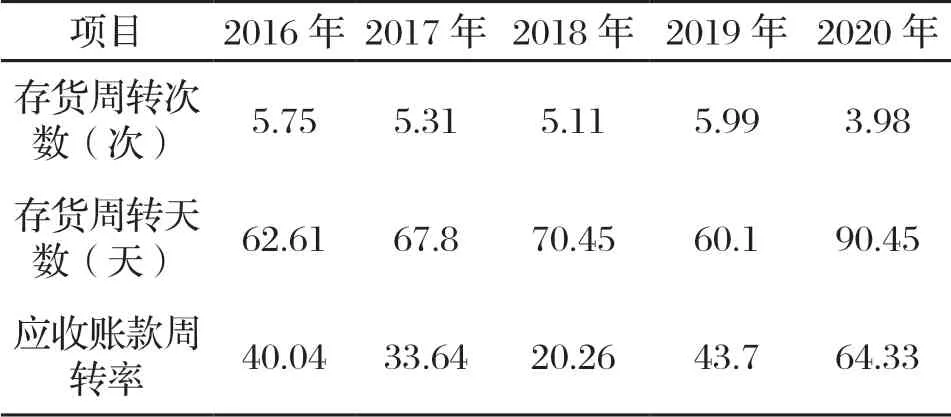
表4 中國飛鶴營運能力指標
2016—2019年中國飛鶴的存貨周轉次數和天數相近;2020年中國飛鶴的存貨周轉次數減少,存貨的周轉天數增加。在通常情況下,企業在一定時間內存貨周轉次數越多越好,存貨的周轉天數越短越好,說明中國飛鶴的存貨周轉速度變慢,占用資金量變大,資金變現速度變慢,存貨庫存壓力變大。
2016—2019年中國飛鶴的應收賬款周轉率為20%~40%;2020年中國飛鶴的應收賬款周轉率為64.33%,大幅度增加。在通常情況下,企業的應收賬款周轉率越高越好,收賬迅速加快,賬齡較短,有利于資金的回收,說明中國飛鶴的應收賬款風險加大,資金占用率高,利用率降低。
1.4 中國飛鶴的盈利能力分析
盈利能力是指企業資金的增值能力。企業的盈利能力通過營業收入利潤率、總資產收益率、凈資產收益率來分析[1]。中國飛鶴盈利能力指標如表5所示。
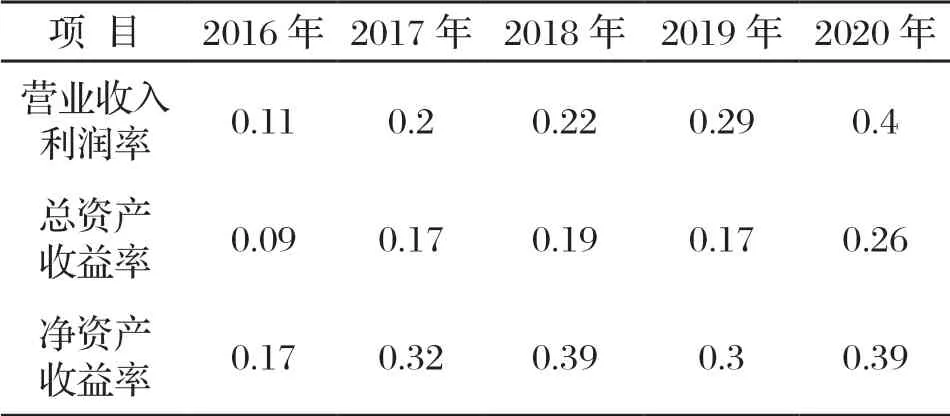
表5 中國飛鶴盈利能力指標
2016—2020年中國飛鶴的營業收入利潤率在逐年增長,說明中國飛鶴獲利水平提升,企業發展良好。
2016年中國飛鶴的總資產收益率0.09,五年中最低;2017—2019年中國飛鶴總資產收益率基本持平,2020年中國飛鶴的總資產收益率0.26,增長較多,說明中國飛鶴的盈利能力增強,發展潛力變大。
2018年中國飛鶴的凈資產收益率在17%;2019—2020年中國飛鶴的凈資產收益率都維持在30%以上,說明中國飛鶴的盈利水平穩定,股東投資所獲得的回報水平得以保障。
1.5 中國飛鶴的發展能力分析
發展能力是指企業擴大規模、壯大實力的潛在能力。企業的發展能力可以通過營業收入增長率、營業利潤增長率、總資產增長率來分析[1]。中國飛鶴發展能力指標如表6所示。
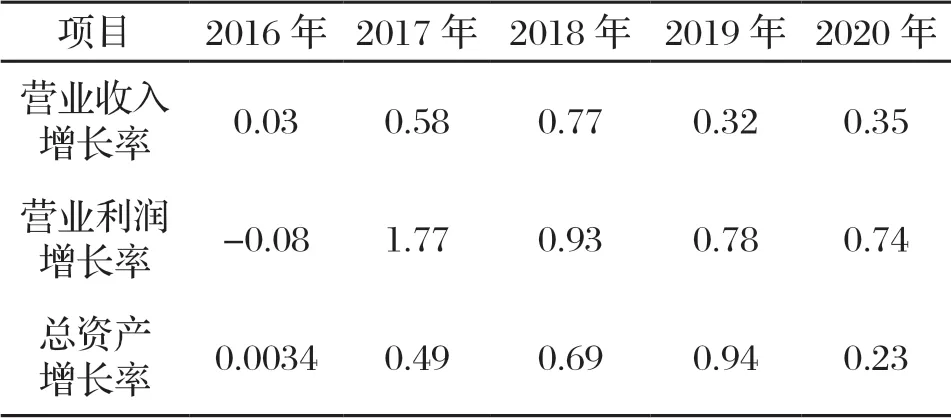
表6 中國飛鶴發展能力指標
2016—2018年中國飛鶴的營業收入增長率從0.03到0.77,增長快速;2019—2020年,中國飛鶴營業收入增長率的增長情況趨于穩定,營業收入增值率越高,說明中國飛鶴發展前景越好。
2016年中國飛鶴的營業利潤增長率為-0.08;2017年中國飛鶴的營業利潤增長率為1.77,說明企業高速發展,營業管理能力大幅度提升;2018—2020年中國飛鶴的營業利潤增長率趨于穩定。營業利潤增長率越高,說明中國飛鶴發展越好。
2016—2019年中國飛鶴的總資產增長率從0.0034發展到0.94,擴張速度飛快;2020年中國飛鶴的總資產增長率為0.23。總資產增長率升高,說明中國飛鶴的資產經營規模擴張速度加快;總資產增長率降低,說明中國飛鶴放緩了擴張速度。
2 借用財務管理促進中國飛鶴發展
2.1 利用財務杠桿,提高資金利用率
一是利用財務杠桿,發揮資金優勢,將資金投入到高端奶粉、綠色奶粉的研究中,以國際標準衡量自己產品的品質,生產出符合國際市場需求的奶粉[3]。二是增強科技創新力和競爭力,對現有奶粉生產鏈條進行升級改造,改進養殖技術和飼養條件,盡量減少加工、運輸等環節,發揮產業鏈與創新鏈作用,利用技術創新體系,加大產品研發力度,生產出高品質的產品[4]。三是發揮國產奶源優勢,堅持綠色低碳環保理念,走綠色奶粉、品質奶粉的發展道路,打造綠色產業模式,用更多的數字化、智能化新技術服務于奶粉生產,提高產品的競爭力。
2.2 加強存貨管理,提高存貨周轉率
首先,應加強產業鏈的布局管理。加快上下游產業鏈的布局,利用“奶農+合作社+企業”的模式,強化奶源基地建設,提高自有奶源的比例,節約生產成本,提高企業競爭力[5]。其次,應進一步加強銷售渠道布局。布局線上線下銷售,線上入駐天貓旗艦店、京東商城、唯品會等電商平臺,線下進入超市和社區[6]。再次,可以借助新媒體淘寶、抖音、快手等直播平臺,打開新的銷售渠道。最后,通過線上線下的有效銜接,節約倉儲、運輸等成本,進而降低存貨總量,提高存貨周轉率。
2.3 抓住政策機遇,提高總資產增長率
一是在全球新冠肺炎疫情的影響下,進口奶粉成本提高,生產周期變長,質量安全事件頻出。為了讓更多的中國人傾向于選擇國產奶粉,應抓準時機,利用國內資源優勢,提高奶粉質量,保障奶粉供應,最大程度保障奶粉市場需求,搶占更多的奶粉市場份額[7]。二是立足國產乳業地域優勢,根據國內消費者需求,走差異化戰略創新路線,在滿足消費者需求的同時,提高市場占有率。三是抓住三胎政策出臺的契機,在增加乳業市場容量的同時,以打造更新鮮、更高品質奶粉為目標,不斷升級轉變,提高市場競爭力,獲取更高地利潤增長空間[8]。
3 小結
通過對中國飛鶴的財務分析,發現其財務管理中資產負債率低、財務杠桿效應發揮不充分、資金利用效率低、存貨多、存貨周轉速度慢、總資產增長率低等問題。提出通過利用財務杠桿提高資金利用效率,加強存貨管理提高存貨周轉率,抓住政策機遇提高總資產增長率等建議,以解決其財務管理問題,進一步提高市場占有率和國際競爭力。
猜你喜歡
發明與創新(2022年30期)2022-10-03 08:40:56
當代水產(2022年5期)2022-06-05 07:55:06
當代水產(2022年3期)2022-04-26 14:27:04
當代水產(2022年2期)2022-04-26 14:25:10
云南畫報(2020年9期)2020-10-27 02:03:26
人大建設(2018年6期)2018-08-16 07:23:10
文理導航·科普童話(2017年5期)2018-02-10 19:42:14
福建輕紡(2017年12期)2017-04-10 12:56:32
現代商貿工業(2016年35期)2016-04-09 06:59:36
小星星·閱讀100分(低年級)(2015年10期)2015-10-22 08:30:04
