黃瓜幼苗下胚軸長度GWAS分析及候選基因挖掘
2020-02-27 03:53:14蔡和序薄凱亮周琪苗晗董邵云顧興芳張圣平
中國農業科學 2020年1期
關鍵詞:分析
蔡和序,薄凱亮,周琪,苗晗,董邵云,顧興芳,張圣平
黃瓜幼苗下胚軸長度GWAS分析及候選基因挖掘
蔡和序,薄凱亮,周琪,苗晗,董邵云,顧興芳,張圣平
(中國農業科學院蔬菜花卉研究所/農業部園藝作物生物學與種質創制重點實驗室,北京 100081)
【】挖掘與黃瓜幼苗下胚軸長度顯著相關的SNP位點及候選基因,為揭示下胚軸長度的遺傳基礎和分子機制提供理論依據,為短下胚軸分子標記輔助選擇育種奠定基礎。以95份黃瓜核心種質為試驗材料,分別于2016年春季、2017年春季、2017年秋季和2018年春季在中國農業科學院南口試驗基地塑料大棚進行種植,在兩葉一心期調查黃瓜幼苗的下胚軸長度;利用Structure 2.3.4軟件分析群體結構,Haploview 軟件分析連鎖不平衡的衰減;基于最優模型對下胚軸長度進行全基因組關聯分析(GWAS),依據關聯SNP位點的LD區間序列,預測與下胚軸長度相關的重要關聯候選基因,并利用熒光定量PCR對預測基因進行表達模式分析。共檢測到8個顯著關聯的位點(),分別位于1、2、3、4、5、6號染色體,其中,等5個位點被重復檢測到兩次以上。通過分析關聯SNP位點的LD區間序列,獲得8個與黃瓜下胚軸長度有關的候選基因,其中既有光形態建成、泛素化、激素信號通路等調控基因,也有調控網絡下游參與細胞生長發育,調節細胞大小,直接調控黃瓜下胚軸長度的基因。多基因在不同黃瓜材料中的有機分布,形成了具有不同下胚軸長度的黃瓜種質。基因表達分析顯示在短下胚軸材料中高表達。在長下胚軸材料中高表達。檢測到等8個與黃瓜下胚軸長度密切關聯的SNP位點,挖掘到等8個調控下胚軸長度的候選基因。
黃瓜;下胚軸長度;全基因組關聯分析;候選基因
0 引言
【研究意義】黃瓜作為一種重要的蔬菜作物,在全球范圍內廣泛栽培。目前黃瓜種植中大量采用工廠化穴盤育苗,單位面積幼苗數量越多即密度越大收益越高,帶來的問題是容易造成幼苗下胚軸徒長。另外,育苗過程中如果遭遇高溫、高濕、弱光等不良環境,也會導致幼苗下胚軸徒長,影響后期豐產性。因此,挖掘黃瓜下胚軸長度的調控位點和基因,開發準確實用的分子標記用于輔助培育短下胚軸品種,對于緩解下胚軸徒長和保證豐產具有重要意義。【前人研究進展】雙子葉植物的下胚軸是連接兩個胚胎葉(子葉)和初生根(胚根)的胚胎干[1]。下胚軸具有相對簡單的結構,是一種非常可塑的器官,受到已知調節細胞伸長的外部和內部因素的強烈影響,例如光照[2-7]、溫度[8]和激素[9-11],在高溫高濕條件下,下胚軸徒長尤為嚴重。Bo等[12]從半野生西雙版納黃瓜中克隆了短下胚軸的基因SHORT HYPOCOTYL1(),該基因編碼SMARCA3染色質重塑因子,它通過與CsHY5進行互作間接調控細胞伸長相關基因的表達,最終影響黃瓜下胚軸的伸長。苗晗等[13]通過下胚軸長度QTL定位檢測到5個下胚軸長度相關QTL,在Chr.5上標記SSR15818和SSR06003之間檢測到3個QTL,總貢獻率高達61%;Chr.6上重復檢出兩個相鄰位點和。全基因組關聯分析(GWAS)是利用自然群體探測物種的遺傳變異,進而挖掘與復雜農藝性狀相關遺傳位點的研究方法,具有不需要構建作圖群體和一次性可同時檢測多個等位基因位點的優勢,近年來在擬南芥[14-15]、玉米[16]、水稻[17]等作物中均有廣泛研究。【本研究切入點】隨著黃瓜基因組測序的完成,利用高通量數據對黃瓜復雜性狀開展GWAS研究越來越便利。QTL定位的方法僅能對在分離群體的親本材料間存在差異的基因效應進行分析,無法在全基因組范圍廣泛挖掘參與下胚軸調控的基因。目前關于黃瓜下胚軸的GWAS研究尚未見報道。【擬解決的關鍵問題】采用GWAS分析鑒定與黃瓜下胚軸長度顯著關聯的SNP,確定控制性狀變異的候選基因。
1 材料與方法
1.1 試驗材料
試驗材料為95份黃瓜核心種質資源,來源于中國農業科學院蔬菜花卉研究所。本核心種質是利用在染色體上均勻分布的23對高多態性的SSR標記,從 3 342份來源于世界各地的黃瓜資源中篩選出來的,具有廣泛的代表性[18]。所有材料均為自然群體,材料編號及來源見電子附表1。
1.2 種植處理
分別于2016年春季、2017年春季、2017年秋季、2018年春季4個時期,將試驗材料在28℃下催芽,出芽后播種于32孔穴盤,于中國農業科學院北京南口試驗基地的溫室育苗。每份材料設置3個重復,每個重復5株,隨機區組排列。
1.3 下胚軸長度調查
黃瓜幼苗長至兩葉一心時,用直尺測量下胚軸長度,記錄原始數據后取平均值。表型數據分析采用SPSS20.0。
1.4 重測序分析與基因分型
整個核心種質群體均完成了全基因組的重測序[18],測序數據見黃瓜基因組網站(http://cucurbitgenomics. org)。過濾掉最小等位基因頻率(MAF)<0.05的變異位點;只保留二態的變異位點。再根據次等位基因頻率(minor allele frequency,MAF)>0.05 和完整度(即非N的SNP位點數占總SNP位點的比例)>0.8對SNP進行篩選,最終得到均勻分布于黃瓜7條染色體上的53 921個高質量且位置唯一的SNP 矩陣用于后續分析。
1.5 群體結構分析與連鎖不平衡分析
群體結構由Structure 2.3.4軟件基于貝葉斯數學模型進行分析,并參照QI等[18]確定最終亞群數目。本試驗為了簡化群體結構復雜性對GWAS結果的影響,K值取為3。用過濾后SNP矩陣進行全基因組連鎖不平衡(linkage disequilibrium,LD)分析[19],分析采用軟件Haploview進行[20]。
1.6 全基因組關聯分析
將苗期調查的表型數據和過濾后構建的高質量SNP矩陣,利用基于R的GAPITR軟件包[21],通過混合線性模型(mixed linear model,MLM)進行關聯分析。利用ggplot2軟件繪制Quantile-Quantile散點圖(QQ plot)[22],并在此基礎上利用QQman繪制曼哈頓圖[23],顯示關聯分析檢測到的與目標性狀顯著相關的標記位點。對95份黃瓜的苗期下胚軸性狀進行全基因組關聯分析。
1.7 候選基因預測
根據GWAS結果獲得的SNP位點,根據序列位置將其定位到黃瓜基因組參考物理圖譜上。基于群體連鎖不平衡分析的LD區間,結合黃瓜基因組網站(http://cucurbitgenomics.org/)基因的功能注釋信息,根據注釋信息找出與性狀相關的候選基因。
1.8 候選基因的表達分析
為了驗證候選基因的準確性,在黃瓜核心種質中選取4個長下胚軸材料(CG9、CG11、CG106、CG112)和4個短下胚軸材料(CG19、CG44、CG54、CG98),在播種后子葉展平、第一片真葉展平、一葉一心、第二片真葉展平、兩葉一心5個時期采集下胚軸。使用日本TaKaRa公司的RNA提取試劑盒(9767)提取總DNA,用1%瓊脂糖凝膠電泳檢測提取得到的RNA質量。用TaKaRa公司的反轉錄試劑盒將提取到的RNA反轉錄成cDNA。用Nanodrop儀器檢測cDNA的濃度及純度,用ddH2O將cDNA稀釋到50 ng?μL-1,用于熒光定量分析。根據預試驗結果選擇黃瓜作為內參基因;使用2-ΔΔCt計算基因的相對表達量。反應體系混合物的配置如表1,PCR反應程序如表2。

表1 熒光定量反應體系

表2 PCR反應程序
2 結果
2.1 表型統計分析
在2016年春季(16S)、2017年春季(17S)、2017年秋季(17A)和2018年春季(18S)4次不同環境下分別對95份黃瓜種質的下胚軸長度進行調查,下胚軸長度差異明顯,統計分析結果見表3。4個批次中材料重復性較好,表現比較一致。長下胚軸材料如CG9、CG11、CG106、CG112等在不同批次中均下胚軸過長,表現為徒長的特性;而短下胚軸材料如CG19、CG44、CG54、CG98等在不同批次中下胚軸長度均較短。4個批次變異系數分別為32.32%、33.27%、26.28%和30.17%,下胚軸長度頻次分布圖具有顯著的正態分布特征(圖1),并且4個批次之間黃瓜下胚軸長度均顯著相關(表4),表明下胚軸長度性狀為典型的數量性狀,采用GWAS分析方法可有效進行該性狀的基因定位。
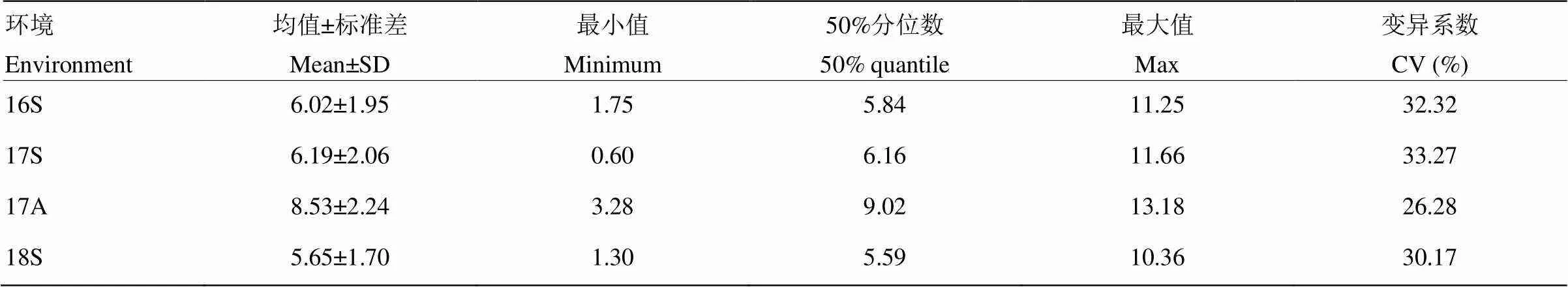
表3 黃瓜下胚軸長度統計分析
16S:2016年春季;17S:2017年春季;17A:2017年秋季;18S:2018年春季。下同
16S: spring of 2016; 17S: spring of 2017; 17A: autumn of 2017; 18S: spring of 2018. The same as below
2.2 群體結構與連鎖不平衡分析
用過濾后的SNP矩陣進行全基因組連鎖不平衡(linkage disequilibrium,LD)分析,當2從0.736衰減到0.368時,所對應的物理距離為17.3 kb(圖2)。因此,黃瓜的全基因組平均 LD為17.3 kb(當2值衰減到其最大值一半時對應的物理長度,即為全基因組平均LD)。

表4 4個批次黃瓜下胚軸長度的相關性分析
** 在0.01 水平(雙側)上顯著相關
** Significantly correlated at 0.01 level (bilateral)

圖1 黃瓜下胚軸長度的頻次分布
2.3 全基因組關聯分析
對黃瓜核心種質苗期下胚軸長度進行全基因組關聯分析(圖3),根據全基因組連鎖不平衡結果,劃分4個批次檢測出的位點所對應的LD區間,對重疊區間進行合并,合并后共8個區間,檢測到8個關聯位點,分別位于第1、2、3、4、5、6號染色體,這些位點可解釋的表型變異率為13.67%— 31.87%。被檢測到3次被檢測到兩次(電子附表2)。說明是在不同季節穩定存在的關聯位點。
2.4 候選基因挖掘
利用已公布的黃瓜基因組測序結果(9930v2),將與下胚軸長度顯著關聯的SNP標記定位到黃瓜基因組上,進而在顯著關聯的SNP位點側翼序列40 kb范圍內提取基因。根據黃瓜基因組的注釋信息,篩選出候選區段內與下胚軸長度相關的同源基因。本研究共篩選出、、、等8個與下胚軸長度相關的候選基因(表5)。其中,、參與植物細胞形態形成,可能直接參與下胚軸的伸長,、可能通過影響光形態發生來調控下胚軸伸長,可能通過蛋白質泛素化來調控下胚軸伸長,、則參與了RNA的加工,可能通過轉錄翻譯的調控來控制下胚軸伸長,則可能通過參與生長素轉運,影響細胞的生長,最終調控下胚軸的伸長。在這些候選基因中,、、、和對應的位點被重復檢測到。
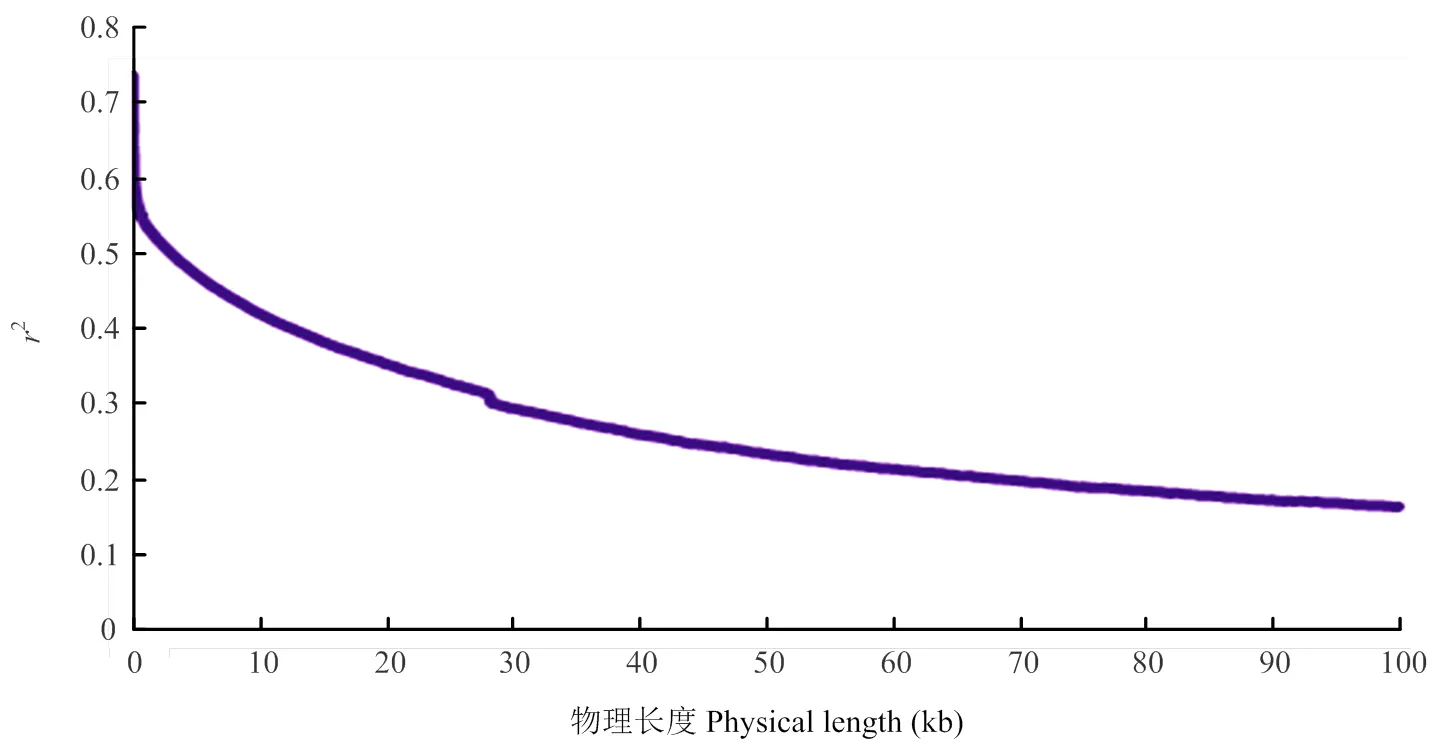
圖2 95份黃瓜全基因組連鎖不平衡衰減

表5 候選基因及注釋信息
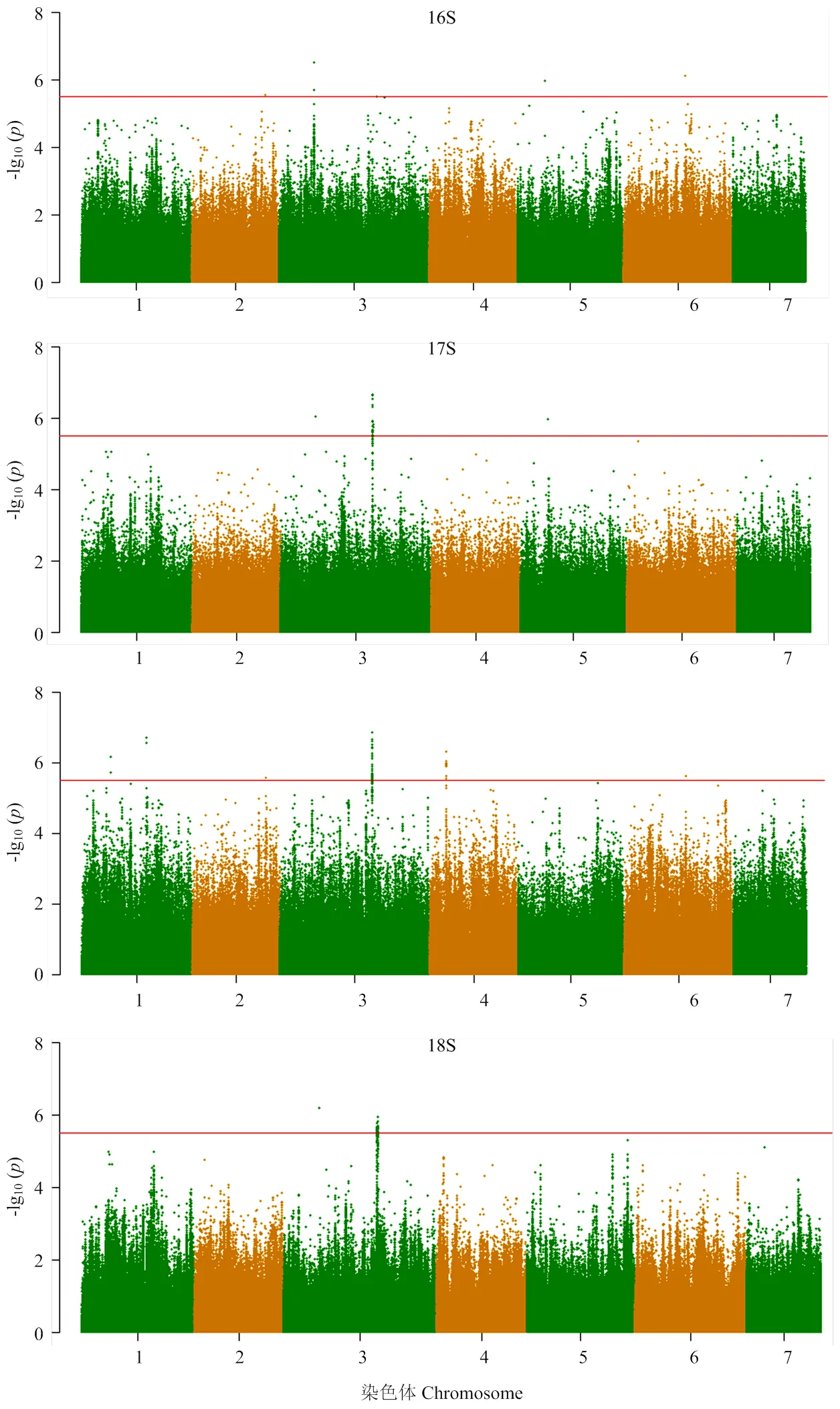
圖3 95份黃瓜核心種質幼苗下胚軸長度的全基因組關聯分析曼哈頓圖
2.5 候選基因的表達模式分析
為了驗證候選基因的準確性,在黃瓜核心種質中選取在4個批次中表現一致的4個長下胚軸材料(CG9、CG11、CG106、CG112)和4個短下胚軸材料(CG19、CG44、CG54、CG98),在播種后子葉展平、第一片真葉展平、一葉一心、第二片真葉展平、兩葉一心5個時期采集下胚軸進行RNA提取,反轉錄后發現其比值均在1.8—2.0,表明提取的RNA質量合格。在瓊脂糖凝膠電泳的膠圖上有兩條清晰可見的條帶,說明RNA有較好的完整性(圖4)。用候選基因和內參的特異性引物分別對其進行熒光定量PCR,其溶解曲線為單峰,說明引物只擴增出單一條帶,具有特異性。
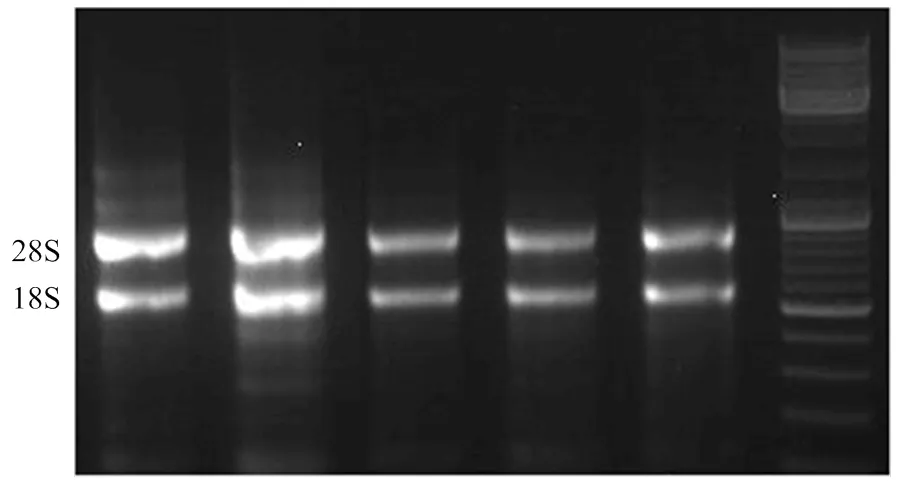
圖4 總RNA電泳檢測
表型調查的數據分析表明,不同材料間下胚軸長度有顯著差異(表6,圖5)。CG9和CG112下胚軸長度最長,過度徒長;CG11和CG106下胚軸長度次之,徒長較重;CG19和CG44輕微徒長;CG54和CG98沒有徒長,幼苗健壯。
熒光定量結果(圖6)顯示,()在CG54和CG98中表達量顯著提高;()在CG19、CG44、CG54和CG98表達量顯著提高;()在CG19和CG98表達量顯著提高;()在CG9和CG11表達量顯著提高;()在CG9、CG11、CG106和CG112中表達量顯著提高;()在CG19和CG54中表達量顯著提高;()僅在CG11和CG106中表達量顯著降低;()在CG9、CG11和CG106表達量顯著降低。總體來看,()、()、()、()、()在短下胚軸材料中高表達。()、()在長下胚軸材料中高表達。

兩葉一心期CG54(左)與CG112(右)

表6 qPCR材料苗期下胚軸長度
時期1:子葉展平;時期2:第一片真葉展平;時期3:一葉一心;時期4:第二片真葉展平;時期5:兩葉一心;表中數據單位均為(cm);不同小寫字母表示不同品種間差異達0.05顯著水平(Duncan’s 法)
Stage 1: the cotyledon stage; Stage 2: the one true leaf stage; Stage 3: the one leaf and one bud stage; Stage 4: the two true leaves stage; Stage 5:the two true leaves and one bud stage; All data units in the table are (cm); Different lowercase letters indicate significant difference between different breeds at 0.05 level (Duncan's method)

CG9、CG11、CG106、CG112均為長下胚軸材料;CG19、CG44、CG54、CG98均為短下胚軸材料;每個候選基因中選取表達量最低的材料,其相對表達量設為1
3 討論
3.1 黃瓜下胚軸長度的變異分析
下胚軸長度是影響黃瓜種苗的重要因素之一,長度過長會影響種苗的運輸、定植后的成活率以及后期單株產量。因此,利用GWAS分析挖掘黃瓜下胚軸長度基因對于黃瓜短下胚軸育種具有重要的價值。本研究選取了來自于東亞、歐洲、美國、西雙版納、印度的黃瓜材料,是從世界各地3 000多份種質中篩選出來的,代表黃瓜75%以上的遺傳變異[18],構建了一個具有豐富下胚軸長度變異的自然群體材料作為試驗樣本,保證了表型數據的多樣性。另外,根據黃瓜實際生產中的條件,在溫室中對核心種質進行4次下胚軸長度鑒定,幼苗長勢和下胚軸長度均表現出明顯差異。篩選出長下胚軸材料CG9、CG11、CG106及CG112,短下胚軸材料CG19、CG44、CG54、CG98等。同時,也有少部分下胚軸長度處于中間的材料在不同季節處理中表現不穩定,可能與處理條件、天氣狀況及溫室內幼苗生物排布方式等有關。
3.2 黃瓜下胚軸長度的QTL定位
本研究通過對95份黃瓜核心種質苗期下胚軸長度的全基因組關聯分析,共檢測出8個顯著關聯的位點,這表明黃瓜的胚軸長度為微效多基因控制。在我國當前的黃瓜保護地生產中,大量采用南瓜作砧木,通過嫁接一定程度上避免了下胚軸徒長的影響。已有研究大部分探究外部環境的生理生化指標對黃瓜下胚軸徒長的影響,例如光強、光質、溫度和激素等[24-26],下胚軸遺傳相關的內容較少,鮮有與之相關的位點和基因。
苗晗等[13]通過QTL定位檢測到5個下胚軸長度相關QTL,在Chr.5上標記SSR15818和SSR06003之間檢測到3個QTL,總貢獻率高達61%;Chr.6上重復檢出兩個相鄰位點和。本研究在5號和6號染色體各兩次重復檢測到1個位點和,但是物理位置相差較大,不是同一個位點。()是生長素信號通路因子,參與生長素轉運調節,可能通過生長素途徑調節細胞的生長發育,進而調控下胚軸的長度。
3.3 黃瓜下胚軸長度候選基因挖掘
光形態發生是一個關鍵的植物發育過程。Bo等[12]從半野生西雙版納黃瓜中克隆了短下胚軸的基因SHORT HYPOCOTYL1(),該基因編碼SMARCA3染色質重塑因子,它通過與CsHY5進行互作間接調控細胞伸長相關基因的表達,最終影響黃瓜下胚軸的伸長。位于3號染色體,本研究在16年春季和17年春季兩次均檢測到與該基因緊密連鎖的SNP(),相距僅50 kb,基本可以確定位點為。熒光定量結果顯示該基因在以西雙版納為代表的短下胚軸材料中表達量顯著下調。在擬南芥中,已證實轉錄因子ELONGATED HYPOCOTYL 5()與組蛋白脫乙酰酶HDA15直接互作,降低組蛋白H4乙酰化水平,參與光形態發生以抑制下胚軸細胞伸長[27-28]。本研究候選基因之一()是HY5在黃瓜中的同源基因,在16年春季和17年秋季被兩次重復檢測到,且在短下胚軸材料中高表達,該基因極有可能通過參與光形態發生調控黃瓜下胚軸的長度。
在17年春季、17年秋季和18年春季3次檢測到了.2,候選基因作為催化亞基和輔助性蛋白質參與組蛋白等多種RNA的加工和降解[29]。基因的表達模式分析顯示該基因在長下胚軸材料中顯著高表達,很有可能作為各種信號通路的下游功能蛋白,影響細胞的生長發育,進而控制下胚軸長度。SCAR 3(,)在擬南芥中則通過調控肌動蛋白細胞骨架的方向來調節細胞的大小[30],主要表現在表皮毛和葉片的細胞中。在黃瓜中極有可能通過影響下胚軸細胞的大小來調控下胚軸的長度。
秋季溫室通常是高溫高濕的育苗環境,下胚軸徒長情況尤為嚴重,因此大量學者研究了高溫下黃瓜幼苗下胚軸長度遺傳情況[6]。高溫情況下黃瓜幼苗下胚軸長度均呈連續變異和正態分布,符合多基因控制的數量性狀遺傳規律;黃瓜幼苗在高溫下的下胚軸長度符合2對加性-顯性-上位性主基因+加性-顯性多基因遺傳模型[8]。本研究在17年秋恰好遇到了高溫高濕環境,絕大部分核心種質下胚軸略有徒長。僅17年秋檢測到的(在擬南芥中的同源基因參與蛋白質泛素化的途徑,與逆境下的反應有關。因此,該位點可能主要與高溫條件下黃瓜下胚軸的伸長有關。
對候選基因的表達分析發現,多次重復檢測到且表達量與表型一致的基因大多數是直接調控細胞的生長發育,進而可能直接調控下胚軸長度的基因。而其他基因多是光形態建成、泛素化、生長素信號通路等轉錄因子,推測應該是感知環境因素的調控基因,只在特定的環境誘導下表達,調控下游細胞發育相關基因,進而調控下胚軸長度。長下胚軸組與短下胚軸組相比,基因表達量趨勢與表型大致相同,但也有一兩個材料不相符,鑒于下胚軸長度是數量性狀,可能是缺失該位點導致,說明這些基因不同程度地參與了下胚軸長度的調控。多基因在不同黃瓜材料中的有機分布,決定了不同的下胚軸長度,形成了具有不同下胚軸長度的黃瓜種質。
4 結論
采用MLM模型分析在4個季節中共檢測到8個與黃瓜下胚軸長度顯著關聯的SNP位點,分別位于1、2、3、4、5、6號染色體。其中兩次以上重復檢測到的位點共5個。通過分析關聯SNP位點的LD區間序列,找到8個與黃瓜下胚軸長度有關的候選基因。調控黃瓜下胚軸長度的候選基因功能多樣,既有光形態建成、泛素化、激素信號通路等調控基因,也有調控網絡下游的功能基因,參與細胞生長發育,調節細胞大小,直接調控黃瓜下胚軸的長度。
[1] LIN Y, SCHIEFELBEIN J. Embryonic control of epidermal cell patterning in the root and hypocotyl of., 2001, 128(19): 3697-3705.
[2] 金正愛, 司龍亭, 李丹丹, 高興. 弱光下黃瓜幼苗葉片下胚軸長的遺傳分析. 江蘇農業科學, 2009(3): 158-161.
JIN Z A, SI L T, LI D D, GAO X. Genetic analysis of hypocotyl length in cucumber seedling leaves under weak light., 2009(3): 158-161.(in Chinese)
[3] 李丹丹, 司龍亭, 羅曉梅, 李濤. 弱光脅迫下黃瓜苗期下胚軸性狀的遺傳分析. 西北農林科技大學學報(自然科學版), 2009, 37(11): 113-119.
LI D D, SI L T, LUO X M, LI T. Genetic analysis of hypocotyl traits in cucumber seedling under low light stress., 2009, 37(11): 113-119. (in Chinese)
[4] 張冠英, 司龍亭, 李丹丹. 弱光脅迫下黃瓜幼苗下胚軸性狀QTL分析. 園藝學報, 2011, 38(2): 295-302.
ZHANG G Y, SI L T, LI D D. QTL analysis for hypocotyl traits of cucumber seedlings under low light stress., 2011, 38(2): 295-302. (in Chinese)
[5] 鄒士成, 李丹丹, 孫博華, 張成鳳, 于思琦. 黃瓜幼苗下胚軸響應弱光脅迫的研究. 安徽農學通報, 2015, 21(19): 29-30.
ZOU S C, LI D D, SUN B H, ZHANG C F, YU S Q. Response of hypocotyl of cucumber seedlings to light stress., 2015, 21(19): 29-30. (in Chinese)
[6] SONG J L, CAO K, HAO Y W, SONG S W, SU W, LIU H C. Hypocotyl elongation is regulated by supplemental blue and red light in cucumber seedling., 2019, 707: 117-125.
[7] 肖蘇琪, 王冰華, 曲梅, 高麗紅. 冬春季節育苗溫室補光光強對黃瓜幼苗質量的影響. 中國蔬菜, 2018(10): 40-45.
XIAO S Q, WANG B H, QU M, GAO L H. Effect of supplementary light intensity on quality of winter-spring cucumber seedling in solar greenhouse., 2018(10): 40-45. (in Chinese)
[8] 張子默, 盧俊成, 齊曉花, 許學文, 陳學好, 徐強. 高溫下黃瓜幼苗下胚軸長度遺傳效應的研究. 分子植物育種, 2019, 17(4): 1326-1332.
ZHANG Z M, LU J C, QI X H, XU X W, CHEN X H, XU Q. Study on genetic effects of hypocotyl length in cucumber seedlings under high temperature., 2019, 17(4): 1326-1332. (in Chinese)
[9] 董春娟, 曹寧, 王玲玲, 張煥欣, 王紅飛, 臺連麗, 尚慶茂. 黃瓜子葉源生長素對下胚軸不定根發生的調控作用. 園藝學報, 2016, 43(10): 1929-1940.
DONG C J, CAO N, WANG L L, ZHANG H X, WANG H F, TAI L L, SHANG Q M. Regulatory roles of cotyledon-generated auxin in adventitious root formation on the hypocotyls of cucumber seedling., 2016, 43(10): 1929-1940. (in Chinese)
[10] LOPEZ-JUEZ E, KOBAYASHI M, SAKURAI A, KAMIYA Y, KENDRICK R E. Phytochrome, gibberellins, and hypocotyl growth (a study using the cucumber (L.) long hypocotyl mutant)., 1995, 107(1): 131-140.
[11] DAN H, IMASEKI H, WASTENEYS G O, KAZAMA H. Ethylene stimulates endoreduplication but inhibits cytokinesis in cucumber hypocotyl epidermis., 2003, 133(4): 1726-1731.
[12] BO K L, WANG H, PAN Y P, BEHERA T K, PANDEY S, WEN C L, WANG Y H, SIMON P W, LI Y H, CHEN J F, WENG Y Q. SHORT HYPOCOTYL 1 encodes a SMARCA3-like chromatin remodeling factor regulating elongation., 2016, 172: 1273-1292.
[13] 苗晗, 顧興芳, 張圣平, 張忠華, 黃三文, 王燁, 方智遠. 黃瓜苗期主要農藝性狀相關QTL定位分析. 園藝學報, 2012, 39(5): 879-887.
MIAO H, GU X F, ZHANG S P, ZHANG Z H, HUANG S W, WANG Y, FANG Z Y. Mapping QTLs for seedling-associated traits in cucumber., 2012, 39(5): 879-887. (in Chinese)
[14] ATWELL S, HUANG Y S, VILHJALMSSON B J, WILLEMS G, HORTON M, LI Y, MENG D, PLATT A, TARONE A M, HU T T, JIANG R, MULIYATI N W, ZHANG X, AMER M A, BAXTER I, BRACHI B, CHORY J, DEAN C, DEBIEU M, de MEAUX J, ECKER J R, FAURE N, KNISKERN J M, JONES J D, MICHAEL T, NEMRI A, ROUX F, SALT D E, TANG C, TODESCO M, TRAW M B, WEIGEL D, MARJORAM P, BOREVITZ J O, BERGELSON J, NORDBORG M. Genome-wide association study of 107 phenotypes ininbred lines., 2010, 465(7298): 627-631.
[15] BRACHI B, MORRIS G P, BOREVITZ J O. Genome-wide association studies in plants: The missing heritability is in the field., 2011, 12(10): 232.
[16] WANG M, YAN J B, ZHAO J R, SONG W, ZHANG X B, XIAO Y N, ZHENG Y L. Genome-wide association study (GWAS) of resistance to head smut in maize., 2012, 196: 125-131.
[17] HUANG X H, WEI X H, SANG T, ZHAO Q A, FENG Q, ZHAO Y, LI C L, ZHU C R, LU T T, ZHANG Z W, LI M, FAN D L, GUO Y L, WANG A H, WANG L, DENG L W, LI W J, LU Y Q, WENG Q J, LIU K Y, HUANG T, ZHOU T Y, JING Y F, LI W, LIN Z, BUCKLER E S, QIAN Q, ZHANG Q F, LI J Y, HAN B. Genome-wide association studies of 14 agronomic traits in rice landraces., 2010, 42(11): 961-967.
[18] QI J J, LIU X, SHEN D, MIAO H, XIE B Y, LI X X, ZENG P, WANG S H, SHANG Y, GU X F, Du Y C, LI Y, LIN T, YUAN J H, YANG X Y, CHEN J F, CHEN H M, XIONG X Y, HUANG K, FEI Z J, MAO L Y, TIAN L, STADLER T, RENNER S S, KAMOUN S, LUCAS W J, ZHANG Z H, HUANG S W. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity., 2013, 45(12): 1510-1515.
[19] YANG J A, LEE S H, GODDARD M E, VISSCHER P M. A tool for genome-wide complex trait analysis., 2011, 88(1): 76-82.
[20] BARRETT J C, FRY B, MALLER J, DALY M J. Haploview: Analysis and visualization of LD and haplotype maps., 2005, 21(2): 263-265.
[21] ZHANG Z W, ERSOZ E, LAI C Q, TODHUNTER R J, TIWARI H K, GORE M A, BRADBURY P J, YU J M, ARNETT D K, ORDOVAS J M, BUCKLER E S. Mixed linear model approach adapted for genome-wide association studies., 2010, 42(4): 355-360.
[22] BRADBURY P J, ZHANG Z W, KROON D E, CASSTEVENS T M, RAMDOSS Y, BUCKLER E S. TASSEL: Software for association mapping of complex traits in diverse samples., 2007, 23(19): 2633-2635.
[23] ZHANG Z W, ERSOZ E, LAI C Q, TODHUNTER R J, TIWARI H K, GORE M A, BRADBURY P J, YU J M, ARNETT D K, ORDOVAS J M, BUCKLER E S. Mixed linear model approach adapted for genome-wide association studies., 2010, 42(4): 355-360.
[24] DAN H, IMASEKI H, WASTENEYS G O, KAZAMA H. Ethylene stimulates endoreduplication but inhibits cytokinesis in cucumber hypocotyl epidermis1., 2003, 133(4): 1726-1731.
[25] ZHAO Q X, YUAN S, WANG X, ZHANG Y L, ZHU H, LU C M. Restoration of mature etiolated cucumber hypocotyl cell wall susceptibility to expansin by pretreatment with fungal pectinases and egta., 2008, 147(4): 1874-1885.
[26] KOEDA S, SATO K, SAITO H, NAGANO A J, YASUGI M, KUDOH H, TANAKA Y. Mutation in the putative ketoacyl-ACP reductase CaKR1 induces loss of pungency in., 2019, 132(1): 65-80.
[27] ZHAO L M, PENG T, CHEN C Y, JI R J, GU D C, LI T T, ZHANG D D, TU Y S, WU K Q, LIU X C. HY5 interacts with the histone deacetylase HDA15 to repress hypocotyl cell elongation in photomorphogenesis., 2019, 180(3): 1450-1466.
[28] CHENGE-ESPINOSA M, CORDOBA E, ROMERO-GUIDO C, TOLEDO-ORTIZ G, LEON P. Shedding light on the methylerythritol phosphate (MEP)-pathway: Long hypocotyl 5 (HY5)/phytochrome- interacting factors (PIFs) transcription factors modulating key limiting steps., 2018, 96(4): 828-841.
[29] RAIJMAKERS R, NOORDMAN Y E, van VENROOIJ W J, PRUIJN G J M. Protein-protein interactions of hCsl4p with other human exosome subunits., 2002, 315(4): 809-818.
[30] BREMBU T, WINGE P, SEEM M, BONES A M. NAPP and PIRP encode subunits of a putative wave regulatory protein complex involved in plant cell morphogenesis., 2004, 16(9): 2335-2349.
GWAS Analysis of Hypocotyl Length and Candidate Gene Mining in Cucumber Seedlings
CAI HeXu, BO KaiLiang, ZHOU Qi, MIAO Han, DONG ShaoYun, GU XingFang, ZHANG ShengPing
(Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing 100081)
【】The aim of this study was to identify SNP loci and candidate genes significantly correlated with cucumber hypocotyl length trait, which could provide a theoretical basis for revealing the genetic basis and molecular mechanism of cucumber hypocotyl length trait, and lay a foundation for marker-assisted selection breeding of cucumber hypocotyl length trait.【】The natural population including 95 cucumber germplasm was employed in this study, and seedlings were grown in the plastic house in Nankou Experimental Field of Chinese Academy of Agricultural Sciences in spring 2016, spring 2017, autumn 2017 and spring 2018, respectively. The hypocotyl length was measured at the two true leaves stage. Structure 2.3.4 software was used to analyze the population structure, and Haploview software was used to analyze the attenuation of linkage imbalance. Then, the whole genome association analysis of hypocotyl length was carried out based on the optimal model. The important candidate genes related to hypocotyl length were predicted according to the LD interval sequence of the associated SNP loci, and the expression pattern of candidate genes were performed by fluorescence quantitative PCR. 【】A total of 8 loci, includingandwere detected on Chr. 1, 2, 3, 4 and 5, respectively. Five of them,and, were detected repeatedly in two or more different environments. By analyzing the LD interval sequences of the associated SNP loci, eight candidate genes,and, were predicted, which were related to cucumber hypocotyl length. Some of the candidate genes involved in regulating plant photomorphogenesis, ubiquitination, and hormone signaling pathway. And some of them were downstream genes regulating cell growth, development and cell size, thus they directly regulated hypocotyl length. Thus, the varied distribution of above genes in different cucumber materials resulted in the different hypocotyl length cucumber germplasm. The organic distribution of polygenes in different cucumber materials formed cucumber germline with different Hypocotyl length. Gene expression analysis showed that,,,andwere highly expressed in short hypocotyl materials andandwere highly expressed in long hypocotyl materials.【】Eight SNP loci linked with hypocotyl length,and, were detected in this study. Eight candidate genes regulating hypocotyl length were predicted, including,,,,,,and.
cucumber; hypocotyl length; genome-wide association study; candidate gene
10.3864/j.issn.0578-1752.2020.01.012

2019-07-24;
2019-09-16
國家自然科學基金(31701929)、中央級公益性科研院所基本科研業務費專項(Y2017PT52)
蔡和序,E-mail:82101172229@caas.cn。薄凱亮,E-mail:bokailiang@caas.cn。蔡和序和薄凱亮為同等貢獻作者。通信作者張圣平,E-mail:zhangshengping@caas.cn。通信作者顧興芳,E-mail:guxingfang@caas.cn
(責任編輯 趙伶俐)
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
