超級電容器電極材料研究進展
2020-03-06 01:52:12李艷梅郝國棟伊廷鋒
化學工業與工程 2020年1期
李艷梅,郝國棟,崔 平,伊廷鋒,4*
(1.安徽工業大學化學與化工學院,安徽 馬鞍山 243002; 2.牡丹江師范學院化學化工學院,黑龍江 牡丹江 157000;3.北京科技大學材料科學與工程學院,北京 100083; 4.東北大學秦皇島分校資源與材料學院,河北 秦皇島 066004)
全球各個領域產業的迅猛發展而帶來的能源短缺和大自然環境遭到破壞成為各個國家亟待解決的難題,這一現狀迫使人類要改變現有能源的使用模式——從依賴傳統的化石燃料轉向大力發展可再生清潔能源[1-2],利用地貌以及氣象條件的優勢,因地制宜地開發綠色、可持續使用的自然資源[3-4]。但是,風能、潮汐能等可再生的清潔能源都會受到季節性和地域性分布的限制,在能源使用方面存在不穩定以及不連續性,很難直接用于工業和日常生活中,故不能滿足社會發展中電力需求的模式[5]。因此,如何存儲這些可再生能源產生的電力并向世界各地按需供應成為人們關注的熱點[6]。目前,在眾多能源存儲與轉化技術中,電化學儲能因具有效率高、可持續性好等優點而具有更廣泛的應用前景。其中,電化學儲能技術中最成熟的就是可充電電池[7-8]。雖然二次電池的使用涉及到生活中大大小小的電子產品,但是其較低功率密度和較短的使用壽命很難滿足當下電動汽車等大型能源設備日益增長的需求[9]。如圖1所示,超級電容器具備輸出功率高(5~30 W·kg-1,大約是鋰離子電池10~100倍)、充電速度快(幾分鐘甚至幾十秒)、循環穩定性好(持續充放電可達到10萬次以上)、安全綠色等優點, 因此,在新一代儲能技術中表現出廣闊的發展前景[10-11]。然而,當代科技的發展需要在不犧牲高功率密度的同時,大大提高能量存儲體系的能量密度和連續穩定的性能。提高能量密度的關鍵在于提高電極材料的比容量和工作電壓,非對稱超級電容器就是一種可行辦法[12-13]。研究者嘗試從各個方面去提高比電容,比如開發出新的電解液以匹配電極材料,深入研究電極材料的儲能機制和失效機制,力求開發出高功率和能量密度儲能體系。本論文擬從各種電極材料的儲能機制和各自的優缺點以及電極材料的設計思路和亟待解決的問題3個方面進行綜述。
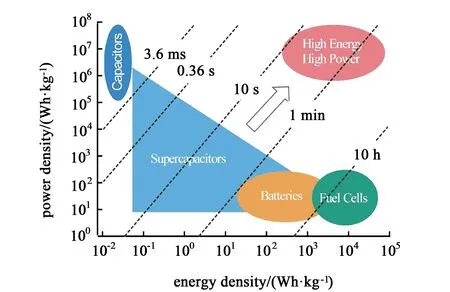
圖1 不同儲能器件的Ragone曲線[12]
1 超級電容器概述
1.1 超級電容器的發展歷史
超級電容器(Supercapacitors,SCs)的發展是發現電荷存儲機制的過程(圖2)。自1740年,萊頓瓶的出現標志著電容器儲能的開始,也是電學研究的重大基礎[14]。1879年,亥姆霍茲發現了金屬電極表面的離子分布狀態,為Gouy-Chapman-Stern提出雙電層模型奠定了基礎。再到1971年,Conway根據RuO2膜的研究結果發現了意義重大的“贗電容”[15-16]。從此,電化學電容器技術跨越了理論到實踐的巨大溝壑,將日漸成熟的技術與混合電動汽車所需的能源動力系統緊密結合[17]。20世紀90年代初期,為了響應時代的需求,世界各國加快了對超級電容器的研究進程,科學家們研發出具有更高能量密度的非對稱電容器和混合型超級電容器[18]。2000年以后,由于超級電容器在儲能領域嶄露頭角,從事電化學電容器研究的公司遍布世界各地,關于超級電容器的研究成果更是層出不窮,各個國家都在以最飽滿的熱情開發和研究不同類型的超級電容器,以供其他領域的能源需求。
1.2 超級電容器儲能原理
1.2.1 雙電層電容器儲能原理
當導電的電極材料浸沒在離子導電的電解質中時,如圖3a)所示,充電時,電解液中的離子(即陽離子和陰離子)與符號相反的同濃度電荷被物理吸附在電極與電解質的界面附近[19-20],則電容存儲在電極材料與電解液之間定向分布的電荷中。放電時,離子從電極表面脫附返回到電解液中。簡言之,充放電過程就是電子和離子在電極和電解液之間定向移動的過程。電容器的比電容的計算如式(1)[21]:
(1)
其中:ε代表介質的介電常數;S代表電極有效比表面積;d代表雙電層的厚度。
雙電層電容器在工作過程中電極與電解液之間沒有電荷的轉移,即不發生氧化還原反應[22]。雙電層電容器的比電容很大程度上取決于電極材料的比表面積及其形貌和表面的官能團性質[23]。雙電層電容器存儲電荷是物理吸附的過程,不涉及到電化學反應,此過程是高度可逆且迅速的(10-8s)[24],因此,雙電層電容器具有更高的功率密度和超長的循環壽命。
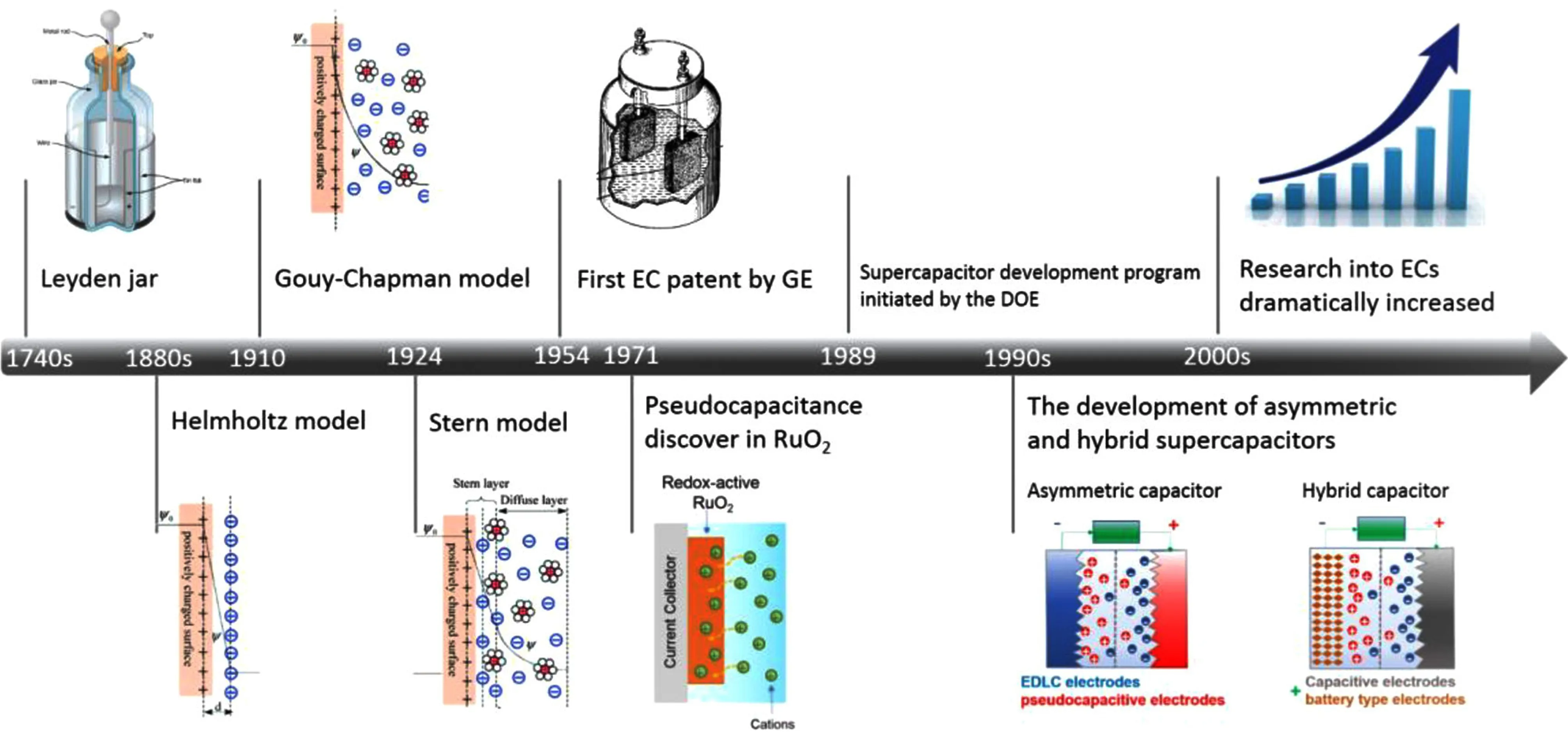
圖2 超級電容器的發展史[14]
1.2.2 贗電容儲能原理
贗電容,又稱法拉第電容,贗電容儲存能量的方式是由法拉第反應過程中的電荷轉移引起的。當充放電達到特定電位時,電荷轉移可以通過欠電位沉積、快速可逆的化學吸附和脫附以及氧化還原反應過程來實現[25]。此過程不僅發生在電極材料表面或近表面,而且電子和離子可以在電極的體系中擴散和傳輸。這一儲能機制中電子轉移會引起電極材料中某種元素的價態變化[26]。如圖3b)~圖3d)所示,不同的儲能機理會產生不同的贗電容特點。圖3b)是欠電位沉積機理示意圖,欠電位沉積是在異種金屬表面上沉積出單原子層或多原子層的現象(例如,Pb2+在Au電極上沉積,H+在Au電極上析出等)。眾所周知,在電位沉積作用下,氫原子吸附在Pt、Rh、Ru和Ir等催化貴金屬上,同時電沉積的金屬陽離子在低于其平衡電位下進行陽離子還原[27-28]。圖3c)為氧化還原贗電容機理示意圖,在氧化還原體系中,利用氧化態的物質(如RuO2、MnO2以及p-型摻雜導電聚合物)和還原態的物質(如RuO2-z(OH)z、MnO2-z(OH)z以及n-型摻雜導電聚合物)之間的電子傳遞。這些反應通常被描述為陽離子在氧化態的物質表面的電化學吸附,在電極和電解質界面之間并伴隨快速且可逆的電子傳遞[29]。RuO2[30]、MnO2[31]以及導電聚合物[32-33]是眾所周知的贗電容器用的電極材料。圖3d)為插層式贗電容電荷存儲示意圖,此機制的核心是在沒有晶體相變的情況下,鋰離子在氧化還原活性物質中嵌入/脫出,其離子嵌入和脫出的速率在時間尺度接近雙電層電容器。典型的插層贗電容材料是T-Nb2O5[34-35],為了保持電極材料的電中性,當鋰離子嵌入材料內部時會引起某種金屬元素價態的變化。
2 超級電容器的電極材料
在超級電容器的各個組成部分中,電極材料起到了不可或缺的作用,其種類和性能極大程度上決定了器件所表現出來的整體性能和應用場所。電極材料主要分為2大類:1)利用材料的表面積和孔道進行物理吸附的雙電層電極材料,如碳基材料[36-38]和碳化物衍生物[39-40];2)利用具有氧化還原活性的贗電容材料,如金屬氧/氫氧化物[41-43]、金屬有機框架[44-45]、導電聚合物[46-47]及金屬硫化物[48-51]。
2.1 碳基材料
隨著各種柔韌性強、可拉伸材料的發展在材料科學上的進步,各種碳基材料廣泛應用于EDLCs,其中,活性炭(AC)、碳纖維、碳納米管(CNT)、石墨烯和碳化物衍生物成為研究熱點[52]。高孔隙率的碳基材料因原料儲量豐富而成本低廉,在化學性質上具有化學和熱穩定性高的特點[53];在物理性質和結構上具有密度小、導電性好、比表面積大等特點,是目前被廣泛研究的電極材料[54-55]。但是在實際服役中,碳材料的表面積并不能完全被利用,孔徑分布及電解液類型會影響雙電層的形成,造成實際比容量只能達到理論容量的10%~20%[56-57]。因此,現在碳材料的研究目標是在不犧牲其功率密度的同時,可控合成分級納米結構,通過調整電極材料的物理/化學性質及修飾碳材料表面來改善碳基材料的能量密度[58-59]。
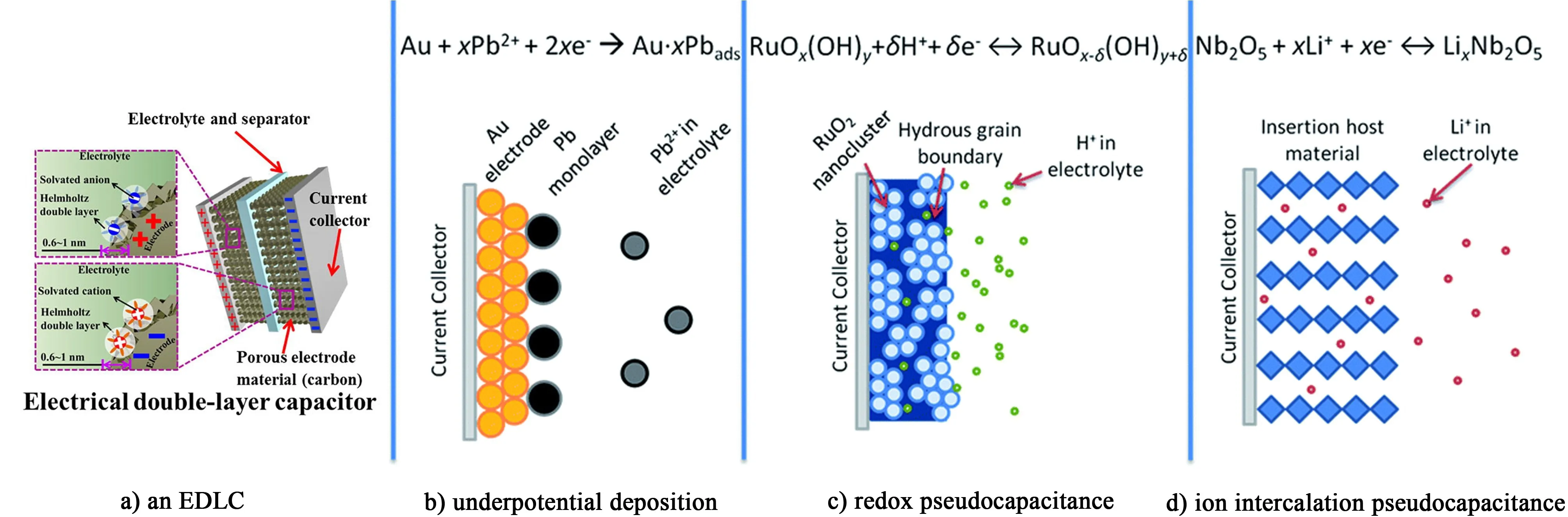
圖3 電荷存儲機制的原理圖:a)雙電層電容器[20];b)欠電位沉積贗電容器;c)氧化還原贗電容器;d)插層贗電容器[25]
He等[60]以煤瀝青成分中的多種芳烴為原料,如圖4a),采用納米ZnO為模板并結合原位KOH活化技術制備了相互連接的三維多孔石墨烯納米囊(GNCs),GNC1/4-AP的比表面積高達1 985 m2·g-1,孔徑尺寸集中分布在0.7~4.0 nm,微孔/超微孔共存使得電極材料具有更好的電化學性能[61]。圖4b)和4c)中可以看出石墨烯納米囊的尺寸大小集中在115~150 nm,其殼的厚度為2.5 nm,該3D GNCs在6 mol/L KOH電解液中,0.05 A·g-1的條件下比容量為277 F·g-1;在20 A·g-1時的比容量為194 F·g-1,經過15 000次循環后的比容量和初始容量相比僅衰減了2.6%,表現了很好的循環穩定性。
金屬有機框架(metal-organic frameworks,MOFs)是一種結構可調控的多孔材料,可以作為模板/前驅體來制備具有高比表面積的多孔碳材料,在超級電容器領域也已經引起了研究者們的廣泛關注[62-63]。Salunkhe等[64]采用ZIF-8直接碳化法制備了一種新型的納米多孔碳材料(NPC)[圖4d)]。如此優異的電化學性能得益于該多孔碳的高比表面積(1 523 m2·g-1)和均勻的粒徑分布[圖4e)和圖4f)]。Guo等[65]用SiO2為模板,在不同溫度下碳化CCl4和乙二胺制備氮摻雜的介孔碳,并且得出適宜碳化溫度為700 ℃。碳化溫度過高,N含量會明顯減小,降低了碳材料與電解液的親和性,比容量會有一定程度的降低。如圖4g)和圖4h)所示,該介孔碳在1 A·g-1的電流密度下,比容量為210 F·g-1,而且,在3 A·g-1的電流密度下持續充放電10 000次,其比容量保持率可達到96.6%。
盡管碳材料已經被廣泛研究且部分商業化,但單一的碳基雙電層電容器的能量密度較低是目前實際生產應用的瓶頸。
2.2 高分子導電聚合物材料
導電聚合物于1976年被發現,是一種常見的導電率和電化學活性比較高的贗電容材料。高分子導電聚合物主要包括聚吡咯(PPy)[66]、聚苯胺(PANI)[67]、聚噻吩(PTh)[68]及其衍生物(PEDOT)[69],這類材料可以采用化學氧化和電化學聚合的方法合成,其存儲能量是通過n(電子)型或p(空穴)型摻雜或去摻雜的氧化還原反應來實現的。因為該反應發生在導電聚合物材料的整個體相中,所以其理論容量比碳基材料高,但電荷進入導電聚合物內部發生反應,由于在充放電過程中伴隨著導電聚合物鏈條持續溶脹和收縮過程,通常導致離子載體擴散能力不足,是其實際應用的主要障礙[70-71]。
高分子導電聚合物的導電性好、比表面積大、易于合成且成本比較低,因此許多研究人員將其運用到電極材料方面,并嘗試了很多方法來改善它們的電化學性能,例如,1)改善形貌和結構,實現納米化;2)采用其它電極材料與導電聚合物進行復合。Wang等[72]采用原位法控制有序的晶須狀聚苯胺在介孔炭表面的生長[圖5b)~5d)],形成“V型”納米孔道,有利于離子的傳輸。在1 mol·L-1的H2SO4電解液中,晶須狀的PANI/介孔碳復合電極的比容量最大值為1 221 F·g-1(0.5 A·g-1);如圖5e)所示,在5 A·g-1的電流密度下循環3 000次后,其比容量僅衰減了5%。因此,為了改善導電聚合物分子鏈結構的有序性、機械穩定性,可采用與其它結構和性質比較穩定的材料進行復合的手段來減小導電聚合物本身體積溶脹、收縮效應,進而提高導電聚合物的比容量和循環穩定性[73]。
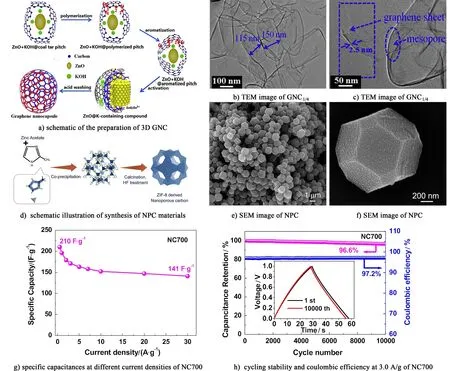
圖4 a)合成3D GNC的機理示意圖; b)和c) GNC1/4的TEM圖[60]; d) NPC電極材料的示意圖; e)和f) NPC的TEM圖[64]; g) NC700的倍率性能; h) NC700在3.0 A·g-1的庫倫效率和循環穩定性[65]
Kong等[74]以碳布為集流體,采用水熱法和化學氧化聚合法制備了NiCo2O4@PPy納米線陣列(NWAs)復合電極[圖5a)]。該結構的優點是具有高導電率的PPy緊密包裹在高比容量的介孔NiCo2O4表面,大大縮短了離子傳輸距離,2種材料的協同作用使得該復合電極具有超高的比容量2 244 F·g-1。而且,如圖5f)和圖5g)所示,當電流密度從1 A·g-1增加到30 A·g-1時,NiCo2O4@PPy NWAs的比容量為初始比容量的60.5%,高于單純的NiCo2O4的倍率容量保持率(52%);即使在10 A·g-1的電流密度下持續充放電10 000次,其容量達到初始容量的82.9%,仍然遠遠高于NiCo2O4NWAs 循環5 000次后的容量保持率78.9%。
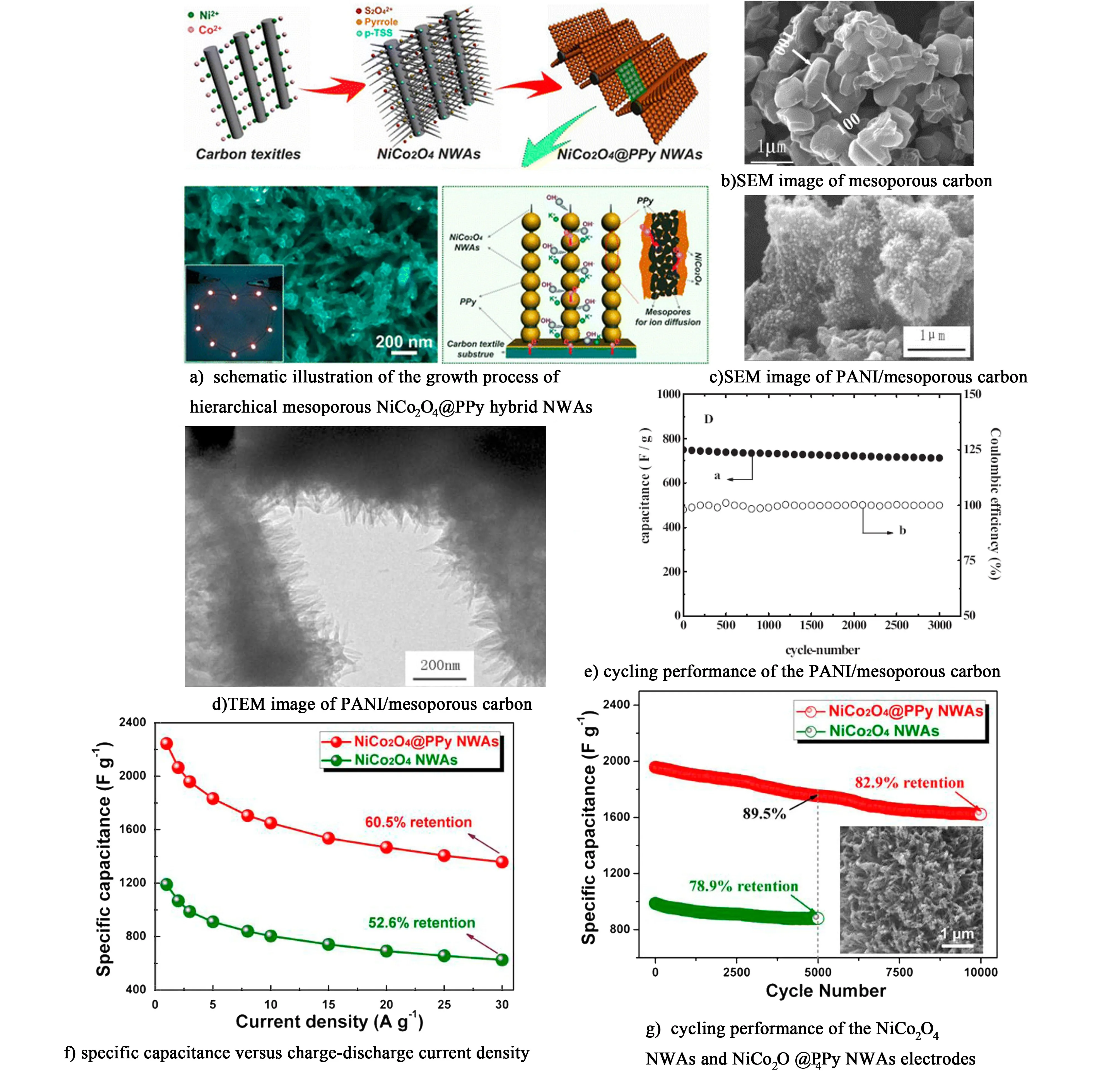
圖5 a)分級介孔NiCo2O4@PPy NWAs的生長機理圖[72]; b)介孔碳的SEM圖; c)和d)介孔炭和PANI/介孔碳復合材料的SEM和TEM圖; e)PANI/介孔碳在5 A·g-1下的循環性能圖; f)和g) NiCo2O4 NWAs和NiCo2O4@PPy NWAs電極的倍率性能和循環性能圖[74]
2.3 金屬氧化物/氫氧化物材料
金屬(氫)氧化物作為典型的贗電容材料,在適當的電壓窗口下發生快速可逆的氧化還原反應但不涉及到相變和結構不可逆的轉化。因此,金屬氧化物作為電極材料的基本要求是電子導電的,或者,金屬元素存在2種或2種以上的價態[75]。金屬氧化物在理論比容量和能量密度方面是碳材料的10~100倍[76],而與導電聚合物相比,其具有穩定的電化學性能。因此,金屬納米材料在電化學能量存儲與轉化領域有更廣闊的發展空間。
RuO2是最先開發的一種贗電容材料,是技術最成熟的金屬氧化物電極材料之一。雖然其在1.2 V電壓窗口下存在3種不同氧化態、優良的電子和離子導電性以及較好的倍率和循環穩定性[77],但RuO2本身有毒且價格昂貴,限制了它在儲能器件中廣泛應用。為了實現可持續發展和降低成本的目標,一些廉價且儲量豐富的單一金屬(MnO2、Co3O4、NiO、V2O5、Fe3O4和CeO2等)和多元金屬氧化物(MnCo2O4、ZnCo2O4和NiMoO4等)“接踵而至”。目前,比較熱門的是Fe、Co、Ni、Mn和Mo等金屬的氧化物及其復合物類電極材料[78-80]。Wei等[81]通過簡單的溶劑熱以及中低溫煅燒的方法合成了分級多孔結構的花瓣粒子狀NiCo2O4/CeO2復合材料[圖6a)~圖6d)],該分級多孔結構的比表面積為113 m2·g-1,有利于電極材料的活性位點暴露在電解液中,增加有效接觸面積。而且,研究表明K4Fe(CN)6作為KOH電解液中的氧化還原類型的添加劑,可以為CeO2及其復合材料提供更適宜的反應場所,顯著改善其電化學性能[82-83]。如圖6e)和圖6f)所示,當K4Fe(CN)6的添加量為0.1 mol/L時,其比容量在5 A·g-1的電流密度下高達2 651.9 F·g-1,同時表現出優異倍率性能和循環穩定性,在10、15、20 A·g-1的電流密度下的比容量分別為2 020.9、1 652.7和1 418.2 F·g-1,而且在10 A·g-1持續充放電5 000次,其容量保持率仍可達到99.6%。
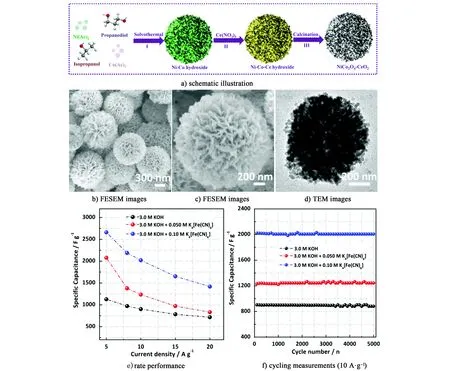
圖6 分級多孔NiCo2O4/CeO2復合材料的a)合成示意圖,b)和c) FESEM圖,d) TEM圖,e)倍率性能,f) 10 A·g-1的循環性能[81]。
2種或以上金屬氧化物復合,不僅可以克服單一氧化物的缺點,而且可以產生顯著的協同效應。以泡沫鎳為集流體,采用改進的水熱法制備得到的MnCo2O4@NiMoO4異質結構的核殼納米線材料[84]。如圖7a)~圖7c)所示,MnCo2O4@NiMoO4的表面包裹約100 nm厚的片狀NiMoO4,提高電極材料的比表面積。MnCo2O4、NiMoO4和MnCo2O4@NiMoO4電極材料的比表面積分別為59.0、87.4和119.2 m2·g-1,很顯然,與MnCo2O4NWAs相比其比表面積增大2倍。因此,MnCo2O4@NiMoO4電極材料具有更高的比容量和更好的倍率性能,電流密度為10 A·g-1時的比容量可達到1 A·g-1時的初始比容量的91%。在5 A·g-1電流密度下進行2 500次循環后,MnCo2O4@NiMoO4仍可提供較高的電容,保持了初始電容的81%,表明其具有很好的循環性能。如此優異的電化學性能得益于多孔的MnCo2O4的核和NiMoO4的殼之間的協同作用以及電活性物質直接生長在導電泡沫鎳基底上,有利于電解質中離子快速地擴散到電極表面。如圖8所示,Yi等[85]通過水熱和化學沉積法合成了花狀的ZnCo2O4/NiO電極材料,采用第一性原理計算得出ZnCo2O4與NiO界面形成了M—O (M=Ni,Co)鍵,這使得改復合材料的結構更加穩定,因此具有良好的循環穩定性,首末十次充放電的形狀沒有明顯變化,說明該電極材料電化學可逆性高。即使在大電流密度30 A·g-1條件下循環充放電4 000次,其比容量與初始容量相比,不僅沒有衰減,而且還有稍微的上升,這是典型的雙金屬氧化物的活化過程導致的。
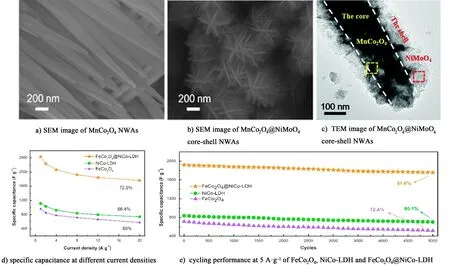
圖7 a) MnCo2O4納米線陣列的SEM圖; b)和c) MnCo2O4@NiMoO4核殼納米線陣列的SEM、TEM圖[85]; FeCo2O4、NiCo-LDH和FeCo2O4@NiCo-LDH電極的d)倍率性能和e) 5 A·g-1的循環穩定性[78]
層狀雙金屬氫氧化物(LDH)具有組成高度可調可控、層間陰離子交換和多功能性質等優點,目前在超級電容器領域大有涉獵,多以復合材料出現,如石墨烯-雙氫氧化物,金屬氧化物-雙氫氧化物等。如圖7d)~圖7e)所示,He等[78]以碳布為導電基底,與NiCo-LDH復合制備出具有核殼結構的FeCo2O4@NiCo-LDH/CC電極材料。其比容量在1 A·g-1的電流密度下高達2 426 F·g-1,同時表現出優異倍率性能和較長的使用壽命,在20 A·g-1的電流密度下的比容量為初始容量的72.5%,而且持續充放電5 000次后,其容量保持率仍高達91.6%。Li等[86]通過采用水熱原位生長的方法制備了花狀的Fe3O4@C@Ni-Al LDH微球[圖9a)];如圖9b)和圖9c)所示,Fe3O4@C表面包裹著一層均勻的Ni-Al LDH,該3層核殼結構的比表面積為72 m2·g-1,且孔徑分布適中,提高電極材料在大倍率下離子快速傳輸的性能。Liang等[87]采用水熱法和磷化反應將NiCo-LDH/NiCoP@NiMn-LDH(NCLP@NiMn-LDH)復合電極材料原位生長在碳布上[圖9d)]。引入NiCoP后,其比容量性能得到很大的提高。在1 A·g-1電流密度下,該電極材料的比容量高達2 318 F·g-1,并且表現出良好的倍率性能[圖9e)]。如圖9f)所示,CoMn-LDH/CF納米墻電極材料在2.1 A·g-1電流密度下的比容量為1 079 F·g-1,而且當電流密度高達42.0 A·g-1時,其容量保持率為82.5%[88]。

圖8 a) ZnCo2O4/NiO材料的理論模型; b) ZnCo2O4/NiO M3電極循環性能(30 A·g-1),內插圖為電極材料在4 000次循環中首末10次充放電曲線圖[85]
2.4 過渡金屬硫化物材料
在多種電極材料中,過渡金屬氧化物/氫氧化物的固有導電性較低,碳材料的理論比容量低,導電聚合物的循環穩定性差等劣勢,嚴重影響了他們在電化學儲能中的大規模應用。最近,過渡金屬硫化物,尤其是單金屬鎳基(Ni2S3和NiS)、鈷基(Co9S8和Co3S4等)和具有立方結構的三元鎳鈷雙金屬硫化物,因為較它們對應的金屬氧化物而言,具有更高的導電性(~100倍)和電化學活性而被廣泛地報道[89]。而且,硫元素的電負性更低,因此,金屬硫化物的結構更加易于調控,體積膨脹效應小,機械穩定性強,有利于電子持續有效的傳輸[90]。金屬硫化物的制備方法簡單,特定的結構和形貌可以通過陰離子交換反應和Kirkendall效應從其對應的金屬氧化物/氫氧化物前體上去構建[91]。然而,目前基于過渡金屬硫化物的電極材料由于其對表面氧化還原反應的依賴性較大,且在大的電流密度下反應動力學速率較慢,其倍率性能以及循環穩定性不佳[92]。因此,設計合理的納米結構,不僅可以改善材料的導電性和原子利用率,還可以縮短電子/離子的傳輸路徑。
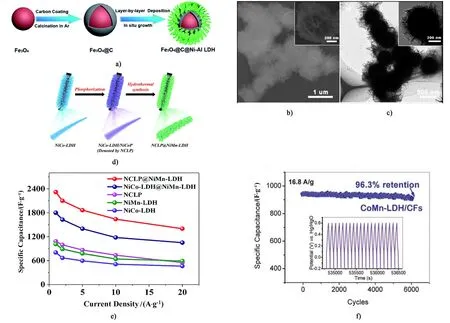
圖9 Fe3O4@C@Ni-Al LDH復合材料的a)合成示意圖; b) SEM; c) TEM圖[86]; d) NCLP@NiMn-LDH復合電極制備流程圖; e)Fe3O4@C@Ni-Al LDH復合物在8 A·g-1的循環穩定性能[87]; f)各種電極的倍率性能[88]
He等[93]以泡沫鎳為導電基底,如圖10a)所示,采用陰離子交換反應(硫化過程)合成了三維多孔網狀結構的Ni3S2/CoNi2S4/NF電極材料,電極材料表面存在脊狀突起增大了其比表面積,該結構和組成優勢賦予其超高的比電容。電流密度2 A·g-1的條件下比容量高達2 435 F·g-1;即使在50 A·g-1的電流密度下,比容量仍可達到1 440 F·g-1[圖10b)],說明該電極材料的倍率性能良好。如此優異的電化學性能歸因于Ni3S2/CoNi2S4/NF電極材料導電性的提高,如圖10c)所示,其電荷轉移電阻遠遠小于其對應的氧化物,促進離子的有效擴散,增強了電化學反應的可逆性。如圖10d)~圖10f)所示,碳包覆的雙殼空心異質結的球形MnCo2S4/CoS1.097(DHMCS@C)電極材料在15 A·g-1的電流密度下連續進行充放電5 000次后其容量可達到初始容量的91.3%,在較大的電流密度下也具有可行的循環穩定性[94]。
Guan等[95]采用連續的離子交換反應制備了洋蔥狀的NiCo2S4顆粒,如圖11a)所示,先制備洋蔥狀的Co4O3顆粒,與S2-交換,變成Co4S3顆粒,再與Ni2+交換變成NiCo2S4顆粒,通過圖11b)~圖11g)可以看到NiCo2S4顆粒的殼是中空的,該結構的比表面積比Co4O3顆粒增大了近2倍,其比容量也進一步得到提高。在電流密度為2和20 A·g-1的條件下,比電容分別為1 061和802 F·g-1,容量保持率僅為75%。
金屬硫化物的比容量在高充放電倍率下保持率較低且在長時間的循環過程中穩定性不理想。這一現象存在是由于硫化物本身容易被氧化,且在持續的循環過程中活性物質易粉化從集流體上脫落。針對這一問題,研究者們嘗試與其他材料進行復合。Chen等[96]通過溫和的水熱法合成了紅毛丹狀NiCo2S4@MnO2復合材料,如圖12a)和圖12d)所示,與MnO2復合后,材料表面由納米變成納米片交聯組裝而成的納米花,該結構具有更大的比表面積和豐富的活性位點,當電流密度從1 A·g-1增加到10 A·g-1時,比電容保持率為78.6%;并在此電流密度下持續循環2 000次后其比容量保持率高達86.8%,表現出良好的電化學性能。Huang等[97]采用簡易的水熱硫化法,以及后續電沉積的方法制備了核殼結構的NiCo2S4@NiO納米線陣列,如圖12e)和圖12f)所示,該電極材料具有很高的面積比電容為12.2 F·cm-2(1 A·cm-2),在20 mA·cm-2的電流密度下的穩定性良好,進行充放電10 000次后比電容的保持率可到達89%,在同一電流密度下其比電容遠遠高于NiCo2S4和NiO單一電極材料。以NiCo2S4@MnO2材料作為正極,活性炭作為負極的全固態電容器,在最高的電流密度測試條件下時的功率密度為0.72 kW·kg-1,比能量密度也可達到10.36 Wh·kg-1,與其他材料的全固態電容器相比,具有競爭性優勢。
綜上所述,超級電容用電極材料主要有如下4類:碳基材料、高分子導電聚合物、金屬(氫)氧化物和金屬硫化物,4種電極材料本身具有優點和不可避免的不足。由碳基材料形成的雙電層電容器一般具有高的功率密度和優異的循環穩定性;但是,其比容量和能量密度比較低。作為贗電容材料之一的導電聚合物,其本身具有良好的電子導電性及較小內阻,但在充放電過程中材料本身易發生體積的膨脹和收縮, 導致其循環穩定性差。而具有較高理論容量的金屬(氫)氧化物本身具有較差的電子傳導性,從而限制了其高功率密度所需的速率性能。而金屬硫化物,它不僅具有較高的理論容量,而且,與其對應的金屬氧化物相比具有更優異的導電性,因此,金屬硫化物成為超級電容器的研究熱點。
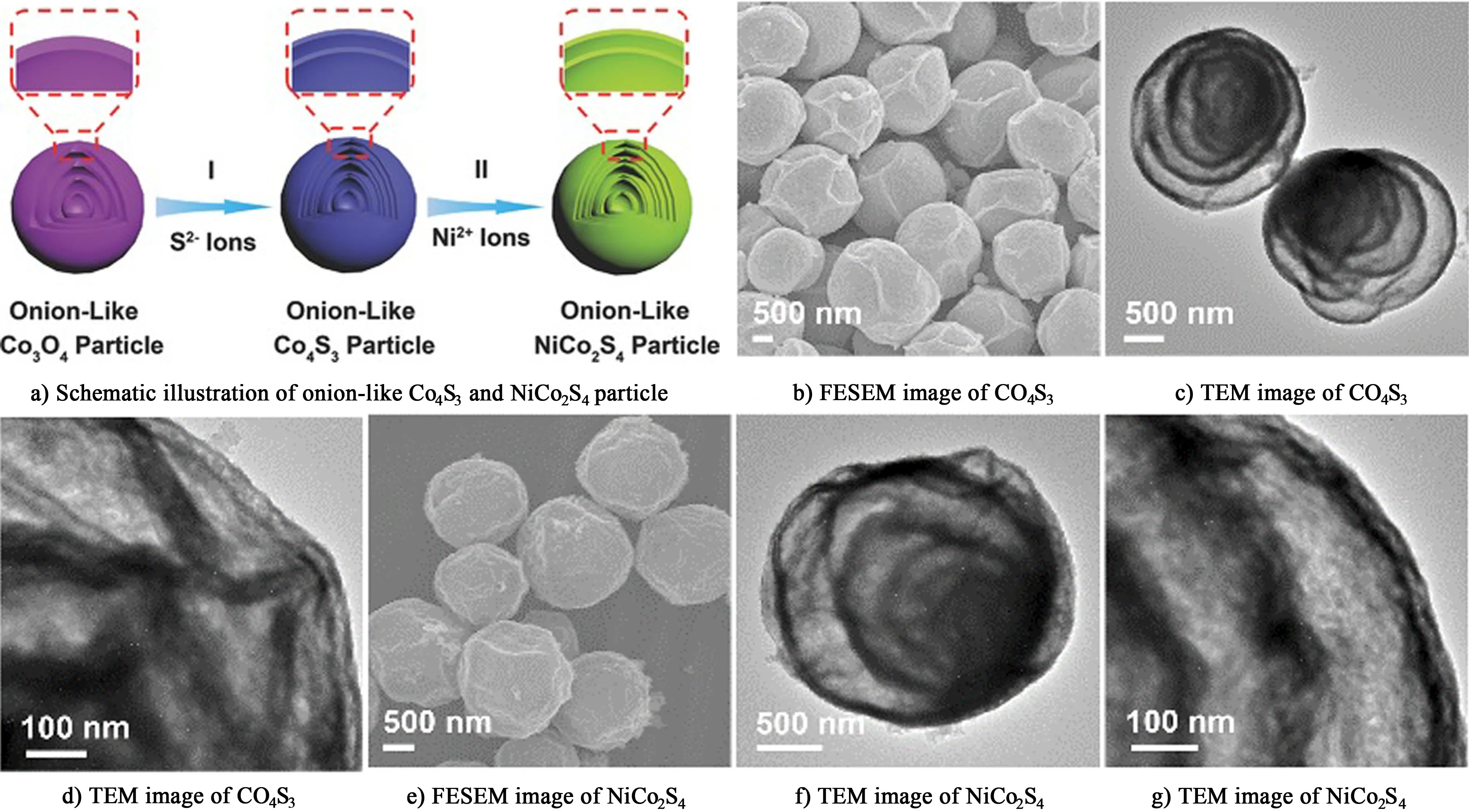
圖11 a)洋蔥狀Co4S3和NiCo2S4的制備機理示意圖; Co4S3的b)FESEM圖; c)和d)TEM圖; NiCo2S4的e)FESEM圖; f)和 g)TEM圖[95]
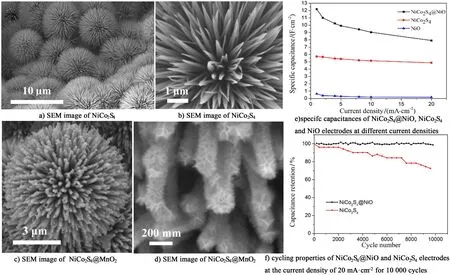
圖12 a),b) NiCo2S4和c),d) NiCo2S4@MnO2的SEM圖[96];NiO、NiCo2S4和NiCo2S4@NiO的e)倍率性能,f) NiCo2S4和NiCo2S4@NiO循環性能[97]
3 結論與展望
總體而言,超級電容器中以氧化還原反應為儲能機制的贗電容器具有更高的理論容量和能量密度,贗電容器電極材料的研究對儲能技術的發展至關重要。但是目前贗電容所具有的能量密度與傳統的電池相比,仍然具有很大的差距。因此,在新型高性能超級電容器用電極材料的研究方面仍具有很大的機遇和挑戰。針對目前電極材料存在的問題,有效解決的辦法主要有3種:1) 材料的合成方面,通過調控溫和反應的條件使電極材料納米化,借此措施增加材料的比表面積,提高電化學活性,從而實現短距離的離子輸運和較快的電子傳導。2) 改善氧化還原反應過程中對電極材料表面依賴性,解決在較大的電流密度下反應動力學緩慢的問題。3) 根據不同種類電極材料的優缺點,研究合成出各種復合材料,力求充分利用不同組分間的協同作用的同時克服單一組分的缺陷。4) 進一步證實形貌決定性能這一理論的相關性,以期指導合成出具有高性能的電極材料。
總的來說,新型超級電容器需要兼具贗電容和雙電層電容的優勢,同時具備優異的倍率性能、高的能量密度和功率密度。雖然在這一領域已經取得很大的進步,但是想要實現這一目標仍然需要研究者們付出更大的努力。
