基于三重四極桿復合線性離子阱質譜法篩查和確證水產品中多種抗生素殘留
2020-03-11 08:44:14朱曉華耿雪冰沈美芳
食品科學 2020年4期
關鍵詞:檢測
孟 勇,王 靜,朱曉華,耿雪冰,楊 總,沈美芳,*
(1.江蘇省淡水水產研究所,江蘇 南京 210017;2.江蘇省水產質量檢測中心,農業農村部漁業產品質量監督檢驗測試中心(南京),江蘇 南京 210017;3. SCIEX亞太應用中心,上海 201206)
近年來,隨著我國水產養殖品種的多元化,養殖規模的集約化,再加上品質、種質以及漁業環境惡化等因素的影響,致使各種水產養殖病害的發生率日趨嚴重,因此為控制水產養殖過程中疫病的發生,藥物的使用已變成常規的養殖輔助手段。目前我國水產養殖常用的抗菌藥物有喹諾酮類、磺胺類、四環素類等,都是廣譜抗菌藥,幾乎對革蘭氏陰性菌、革蘭氏陽性菌、克雷伯氏菌屬、沙門氏菌屬、變形桿菌屬、某些支原體和衣原體等都具有較強的抑菌作用[1-3]。但由于水產養殖的特殊性及缺乏相應的科學指導,養殖業者在水產養殖過程中為保證藥效常常超量或者違規使用藥物,藥物品種越來越復雜,對水產品中藥物殘留篩查和確診技術的要求也越來越高。
目前,國內外對水產品中抗生素殘留的研究日趨重視,檢測方法主要有酶聯免疫法[4-6]、液相色譜法[7-9]、液相色譜-串聯質譜法[10-13]、高分辨質譜等[14-15]。酶聯免疫法具有操作簡便、檢測成本低等優點,但定量不夠準確,容易出現假陽性。液相色譜法具有分析效率高、定量準確等優點,由于抗生素藥物種類繁多和樣品基質的復雜性,其在多種藥物的同時篩查和藥物的定性方面具有一定的局限性。高分辨質譜因為其質量數準確,定性能力強,但該儀器定量能力弱,價格相對昂貴,普及率較低。本研究應用液相色譜-三重四極桿復合線性離子阱質譜技術,采用分段時間多反應監測、信息關聯采集、增強子離子掃描、譜庫檢索的檢測模式[16-18]。其原理是根據各個化合物的保留時間,在其設定的時間窗口內,當多反應監測信號超過設定的閾值時,多反應監測和增強子離子掃描會同時觸發,得到該化合物的二級質譜碎片的全譜信息。并且實現了在優于常規三重四極桿液相色譜-質譜聯用儀利用2 對子離子的采集信息進行定性定量的基礎上,結合建立的增強子離子掃描譜庫對比功能,對可疑色譜峰通過二級譜庫檢索進一步確證。目前水產品中藥物殘留的篩查和確證方法主要還是利用高分辨質譜檢測技術[19-21],但是基于三重四極桿復合線性離子阱質譜法針對水產品殘留確證技術的報道很少[22]。因此,本方法擬實現同時對水產品中喹諾酮類、磺胺類、四環素類等多種抗生素殘留進行快速篩查和確證,為保障我國水產品質量安全和風險監測提供有效手段。
1 材料與方法
1.1 材料與試劑
甲醇、乙腈(均為色譜純) 德國Merck公司;乙酸銨、甲酸、乙酸(均為色譜純) 美國ROE公司;乙二胺四乙酸二鈉(分析純) 國藥集團化學試劑有限公司;土霉素、金霉素、四環素、氘代恩諾沙星、氘代磺胺鄰二甲氧嘧啶、氘代土霉素標準品 德國Dr.Ehrenstorfer公司;喹諾酮類(16 種)和磺胺類(19 種)標準品(質量濃度為100 mg/L,純度均大于98.3%) 天津阿爾塔科技有限公司;實驗用水為Millipore系統超純水。
1.2 儀器與設備
QTRAP 4500液相色譜-質譜聯用儀 美國SCIEX公司;T18勻漿機 德國IKA公司;AllegraTM離心機美國Beckman公司;Turbo Vap濃縮工作站 瑞典Biotage公司。
1.3 方法
1.3.1 混合標準溶液配制
粉末狀標準品均用甲醇配制成100 mg/L的標準儲備液。從質量濃度為100 mg/L的標準儲備液中各取100 μL,用甲醇定容至10 mL,制備質量濃度為1.0 mg/L的混合標準儲備液,于-18 ℃保存備用。實驗中再用0.1%甲酸-乙腈(9:1,V/V)溶液作為溶劑,逐級稀釋,配制成不同質量濃度的混合標準使用液待用。
氘代恩諾沙星、氘代磺胺鄰二甲氧嘧啶、氘代土霉素標準品分別用1.0%甲酸-甲醇溶液配制成100 mg/L的內標儲備液。從上述質量濃度為100 mg/L的內標儲備液中各取100 μL,用0.1%甲酸-乙腈(9:1,V/V)溶液作為溶劑,制備質量濃度為1.0 mg/L的混合內標溶液。
1.3.2 樣品前處理
稱取2.0 g均質樣品于50 mL離心管中,加入1.0 mg/L混合內標溶液50 μL,渦旋混合30 s,避光放置5 min。再加入10 mL 1%甲酸-乙腈溶液和0.1 g乙二胺四乙酸二鈉,渦旋振蕩1 min,超聲提取3 min,8 000 r/m離心5 min,取上清液。殘渣中加入10 mL 1%甲酸-乙腈溶液,并用玻璃棒搗碎,重復提取1 次,合并2 次提取液,搖勻備用。提取液于40 ℃氮氣條件下吹干,殘渣用1 mL的0.1%甲酸-乙腈(9:1,V/V)溶液溶解,渦旋振蕩30 s,加入2 mL正己烷,渦旋振蕩30 s,4 000 r/min離心5 min,移取下層溶液于1.5 mL高速離心管中,12 000 r/min離心5 min,取上清液經0.22 μm濾膜后上機測定。
1.3.3 液相色譜-質譜條件
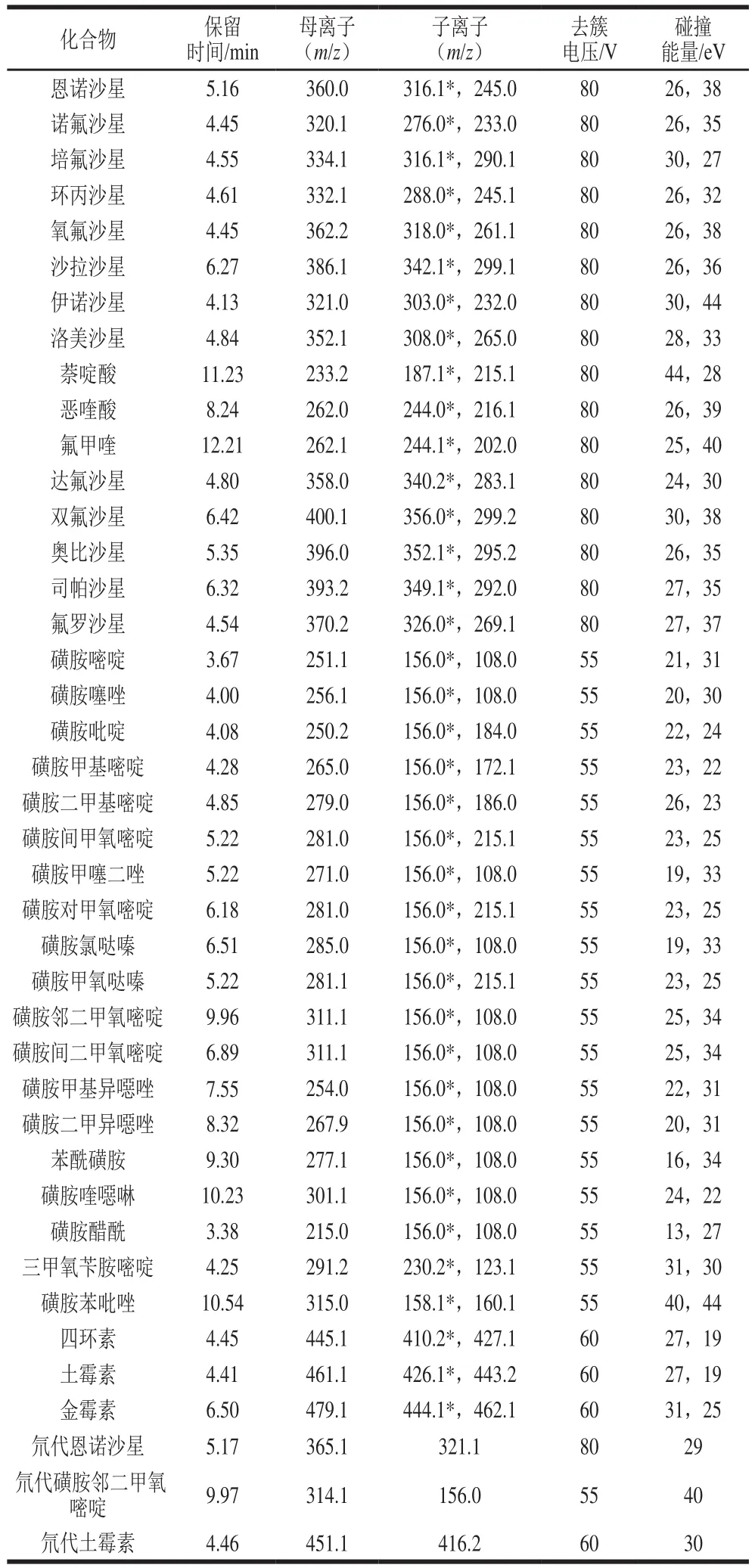
表1 多種抗生素和3 種內標化合物質譜條件參數Table 1 Mass spectrometric parameters for multiple antibiotics and three internal standards
Agilent Poroshell EC-C18色譜柱(100 mmh2.1 mm,2.7 μm);流動相為0.1%甲酸溶液(A)和0.1%甲酸-乙腈溶液(B);柱溫35 ℃;流速0.3 mL/min;進樣量10 μL。梯度洗脫程序:0~1 min,97% A;1~2 min,97%~85% A;2~9 min,85%~75% A;9~15 min,75%~45%A;15~16 min,45%~97% A;16~20 min,97% A。電噴霧離子源正離子模式;多反應監測-信息關聯采集-增強離子掃描檢測模式;離子化電壓5 500 V;離子源溫度500 ℃;氣簾氣壓力0.21 MPa;霧化氣壓力0.41 MPa;輔助加熱氣壓力0.41 MPa。具體質譜參數見表1。
1.4 數據處理
采用SigmaPlot 11.0處理數據統計及圖表繪制。
2 結果與分析
2.1 前處理條件優化
目前磺胺類、喹諾酮類、四環素類藥物的提取劑主要有:乙腈[23]溶液、三氯乙酸溶液[24]、甲醇溶液[25]、磷酸鹽緩沖液[26]等。本實驗考察提取溶劑種類、提取方式、提取用量等因素對提取效果的影響,結果發現乙腈作為提取劑具有較好提取效果和去蛋白特性,也更有利于后續的凈化處理,優于其他提取劑。質譜檢測時采用正離子模式,因此加入適當的酸性溶液有助于提取分析物的電離和提取,但四環素類藥物的回收率偏低,不足40%。參考相關文獻[12],使用1%甲酸-乙腈溶液作為提取液,加入0.1 g乙二胺四乙酸二鈉,四環素類化合物的回收率明顯增加,其他化合物的回收率在70%~110%之間,從而保證了所有分析目標物的提取效率。
樣品凈化主要方式有液液萃取[11]、QuEChERS[14]、固相萃取[13,26]、分散固相萃取[27]等。本實驗加入了QuEChERS常用的無水NaSO4或無水MgSO4等凈化劑,可能是因為化合物的水溶性或者產生的放熱反應,對部分抗生素特別是喹諾酮類和四環素類化合物的回收率有較大影響。喹諾酮和磺胺類藥物結構中含有羧基、堿性氮原子或氨基,顯示出酸堿兩性。HLB固相萃取柱是親水-親脂平衡型柱,對極性和非極性化合物均有很好的保留,在獸藥殘留檢測中得到廣泛應用[29],但固相萃取操作過程較為繁瑣、耗時長、成本較高,批次間實驗結果重復性較差。因為本方法的儀器檢測條件對分析目標物具有很高的特異性選擇,檢測圖譜中較少雜質峰干擾,正己烷去脂能夠滿足檢測要求,從而降低了檢測成本,縮短了前處理時間。
2.2 色譜條件優化
比較Cortecs C18(100 mmh3.0 mm,2.7 μm)、Kinetex C18(100 mmh2.1 mm,2.6 μm)和Agilent Poroshell EC-C18(100 mmh2.1 mm,2.7 μm)3 種色譜柱分離多種抗生素的分離效果。結果表明,3 種色譜柱均能將38 種抗生素化合物分離,但Cortecs C18各色譜峰出峰時間較長,最遲出峰的氟甲喹峰形拖尾較為嚴重,四環素類抗生素在Agilent Poroshell EC-C18色譜柱的響應值相對更高,各分析目標物分離效率高,穩定性好,色譜峰形優異。
分別比較甲醇和乙腈作為本實驗流動相的有機相對各分析目標物的分離效果影響。結果表明甲醇作為流動相時,離子化效率要優于乙腈,相應值更高,但乙腈的基線噪音要優于甲醇,分析目標物的靈敏度相對更高。因此,相較而言本實驗選擇乙腈作為強洗脫流動相。
本實驗分別選取水-乙腈、0.1%甲酸-乙腈溶液、含0.1%甲酸的5 mmol/L甲酸銨-乙腈溶液、含0.1%甲酸的5 mmol/L乙酸銨-乙腈溶液作為流動相,比較不同流動相體系的洗脫效果。結果表明甲酸能夠增加各種化合物在電噴霧離子源正離子模式下的離子化效率,峰形對稱尖銳;乙酸銨雖然能夠增加部分化合物的離子化效率,但是峰形變寬,質譜圖中部分色譜峰會出現毛刺;甲酸銨會造成部分化合物分離效果不佳,后出峰的化合物拖尾嚴重,保留時間延長。故選擇0.1%甲酸-乙腈溶液作為流動相。
2.3 質譜條件優化
在正離子模式下,38 種抗生素及3 種內標均可獲得較高的[M+H]+準分子離子峰[29]。本實驗分別通過對各化合物碰撞選擇離子、碰撞能量和去簇電壓等質譜參數進行優化,選取經碰撞后豐度較高的2 個子離子作為定量和定性離子,最終選擇確定的掃描時間窗口、母離子、子離子和碰撞能量等參數(表1)。磺胺間二甲氧嘧啶和磺胺鄰二甲氧嘧啶作為同分異構體,在本實驗的檢測條件下能夠完全相互分離,保留時間分別為9.96 min和6.89 min。按優化條件進行測定,見圖1。
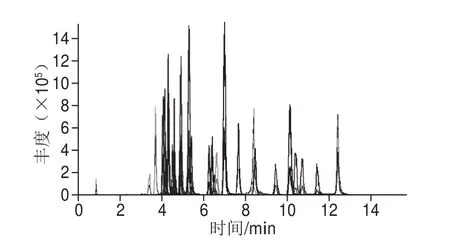
圖1 質量濃度均為20 μg/L的38 種抗生素混合標準溶液在多反應監測模式下的總離子流圖Fig. 1 Total ion current chromatogram for a mixed standard solution of the 38 antibiotics (20 μg/L) in MRM mode
王志杰等[11]研究33 種喹諾酮和磺胺類藥物殘留檢測以及郭萌萌等[12]研究的23 種漁藥殘留檢測等漁藥多殘留分析液相色譜-質譜聯用技術,均采用常規的多反應監測模式掃描,因為監測的化合物較多,為保證每個化合物色譜峰采集的點數足夠,分配的駐留時間設定非常小。通常情況下,每個色譜峰需要采集12 個點,才能使其具有良好的峰形和重復性,駐留時間對化合物的峰形影響很大[30]。在實際檢測過程中存在水產品各種復雜基質的干擾,由于駐留時間很短,往往很難保證化合物能夠采集足夠的點數,保證檢測結果的準確定性和定量。因此,本實驗采用多反應監測-信息關聯采集-增強子離子及分段時間掃描模式,利用每種化合物保留時間分別確定每個化合物的掃描窗口,在設定的掃描窗口只監測該化合物的定性、定量離子,從而保證每個化合物所分配的掃描窗口得到足夠駐留時間。同時當多反應監測信號超過設定的閾值時,Q3會自動切換為線性離子阱,并對該多反應監測通道執行增強子離子,獲得二級質譜圖,從而對多反應監測的檢測判定予以補充。
2.4 化合物譜庫的建立及檢索
常規的串聯四極桿質譜只能得到多反應監測的2 對離子的色譜峰,定性和定量僅靠兩對離子的豐度比確定。本實驗采用三重四極桿復合線性離子阱質譜儀,并采用多反應監測-信息關聯采集-增強子離子掃描模式,在多反應監測的基礎上還可以采集增強型二級質譜圖。另外,該譜庫是開放性的,可以在原有譜庫的基礎上,采集高、中、低(50、40、30 eV)3 種碰撞能量下添加標樣后的實際樣品二級質譜圖,從而對樣品基質條件下的增強子離子圖進行補充,提高匹配率。本方法實現了一次進樣,可以同時得到多反應監測的色譜圖和高靈敏度的增強子離子圖,并能夠通過保留時間、定量離子、輔助定性離子及二級質譜譜庫的增強子離子譜庫比對進行定性、定量分析,相較于常規三重四極桿液相色譜-質譜聯用儀的檢測,可有效降低假陽性或假陰性樣品結果。
2.5 方法學驗證
取質量濃度分別為1.0、2.0、5.0、10.0、50.0、100.0、200.0 μg/L一系列混合標準溶液按1.3.3節方法上機測定,以目標物的質量濃度x(μg/L)為橫坐標,定量離子和相應內標離子峰面積的比值y為縱坐標繪制工作曲線。結果顯示,恩諾沙星等16 種喹諾酮類化合物的線性范圍為1.0~200.0 μg/L;磺胺嘧啶等19 種磺胺類化合物的線性范圍為1.0~200.0 μg/L;四環素等3 種四環素類化合物的線性范圍為2.0~200.0 μg/L,所有化合物的線性相關系數均大于0.99。以3 倍信噪比確定化合物的檢出限。選用陰性草魚作為空白基質,分別添加3 個質量濃度水平的加標樣品,其中最低添加量為方法定量限,每個水平做6 個平行樣品,按照1.3.2節進行樣品前處理、以1.3.3節測定條件進行檢測,結果見表2。結果表明38 種抗生素在3 個不同添加水平的平均回收率為63.5%~117.3%,相對標準偏差為2.6%~14.8%,檢出限為0.5~2.0 μg/kg,定量限為1.0~5.0 μg/kg。

表2 多種抗生素的檢出限、回收率和相對標準偏差(n=6)Table 2 Limits of detection, recoveries and relative standard deviations of multiple antibiotics (n= 6)
2.6 實際水產品中38 種抗生素殘留的分析檢測結果
將該方法用于池塘和湖泊養殖河蟹、草魚、鳙魚、鯉魚、鯽魚、鱸魚、鰱魚、斑點叉尾鮰、烏鱧、鱖魚、克氏原螯蝦、南美白對蝦等13 個品種289 個樣品中抗生素的快速篩查分析。結果發現有10 個草魚、8 個鯉魚、7 個鯽魚、5 個鱸魚、2 個烏鱧、2 個克氏原螯蝦、2 個斑點叉尾鮰共36 個樣品篩查出含有恩諾沙星殘留;3 個鱸魚、2 個草魚、2 個鯽魚、1 個烏鱧共8 個樣品篩查出含有環丙沙星殘留;1 個鯽魚、1 個鳊魚篩查出含有氧氟沙星殘留,1 個鯽魚篩查出含有磺胺間甲氧嘧啶殘留;2 個鳊魚分別篩查出含有磺胺嘧啶和磺胺二甲基嘧啶。
以上結果中有2 個喹諾酮類陽性可疑樣品,通過查看未知化合物增強型二級全掃譜圖,并與譜庫中的二級全掃譜庫進行匹配,結果見圖2、表3。可疑的草魚樣品中恩諾沙星正向匹配率為88.91%,反相匹配率為89.23%,平均匹配率為84.72%。另一個可疑克氏原螯蝦樣品中恩諾沙星正向匹配率為90.0%,反相匹配率為45.1%,平均匹配率為40.3%。分析原因是克氏原螯蝦的基質成分和譜庫中的基質成分有所差異,造成了反相匹配率較低,因此通過在克氏原螯蝦空白樣本中添加標準溶液,采集其相關數據添加到譜庫后重新進行匹配,反相匹配率提高為89.7%,平均匹配率為85.1%。

圖2 陽性可疑樣品的增強子離子掃描譜圖(A)與譜庫中的恩諾沙星標準譜圖(B)的對比Fig. 2 Comparison of EPI spectrum of a doubtful positive sample (A)and the library spectrum of enrof l oxacin (B)
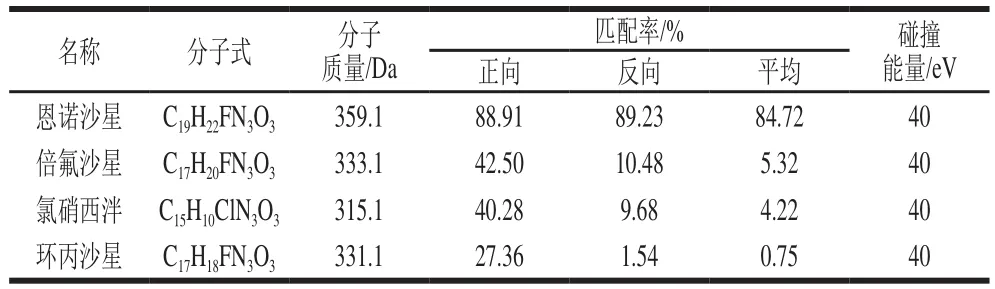
表3 陽性可疑樣品的增強子離子掃描與譜庫匹配及低匹配度篩選結果Table 3 Results of searching for EPI chromatogram of a doubtful positive sample in the library
3 結 論
本實驗采用三重四極桿復合線性離子肼質譜法建立水產品中38 種抗生素的篩查與鑒定方法,結合實驗室自建的二級全掃譜庫可對陽性可疑樣品進行鑒定。通過分段時間多反應監測-信息關聯采集-增強子離子掃描的檢測模式,一次進樣即可同時得到多反應監測色譜圖和高靈敏度的二級碎片質譜圖,通過譜庫檢索排除假陽性或假陰性結果,相較于常規的漁藥多殘留液相色譜-質譜聯用儀采用多反應監測檢測技術,更加具有優勢。綜上所述,該方法分析速度快、靈敏度高、篩查范圍廣,適用于水產品中抗生素殘留的篩查與鑒定,為滿足實驗室進行日常檢驗和風險監測提供了一種有力的技術手段。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
