基于配位鍵交聯的自修復彈性體的合成及表征
2020-04-02 12:04:42柏至偉張治斌張雅娟汪濟奎
功能高分子學報 2020年2期
關鍵詞:力學性能
柏至偉, 張治斌, 張雅娟, 汪濟奎
(華東理工大學材料科學與工程學院,超細材料制備和應用重點實驗室,上海 200237)
自修復材料是一類能夠自發地或者在外界環境刺激下對自身物理缺陷進行修復的智能材料,因其能夠延長材料的使用周期,提高材料的可靠性和減少材料的浪費而引起廣泛關注[1-3]。
有機硅彈性體因其具有疏水性、耐化學腐蝕性、耐高低溫性以及良好的生物相容性而被廣泛應用于電子設備、汽車、化學工程和航空航天等領域[4-6]。有機硅彈性體通過縮聚、共聚和硅氫加成等方式進行交聯可獲得優異的力學性能,然而這些交聯過程大部分是以不可逆的[7,8],所以在有機硅彈性體受到機械損壞后無法進行自我修復和回收利用,造成了材料的極大浪費和環境污染。因此設計具有自修復性能的有機硅彈性體,符合未來社會對環境保護,資源可持續利用的需求。在材料中引入動態化學鍵,如酰腙鍵[9]、硼氧鍵、二硫鍵[10,11]、配位鍵、氫鍵等,或通過Diels-Alder[12,13]反應、酯交換等可逆反應,可以使材料具有自修復性。自修復的過程需要在一定的條件下進行,其中配位鍵實現自修復的條件較為溫和。在之前的研究當中,Li 等[14]通過酰氯吡啶將吡啶官能團接入聚二甲基硅氧烷主鏈,得到具有自修復能力的有機硅彈性體,其拉伸強度為0.25 MPa。Jia 等[15]通過將1,2,4-均三唑共聚進入聚二甲基硅氧烷主鏈實現有機硅彈性體的自修復,其拉伸強度為0.4 MPa。Liu 等[16]將吡啶-2-甲醛通過席夫堿反應接枝到氨基硅油側鏈上,以鈷離子交聯制備自修復有機硅彈性體,拉伸強度為0.45 MPa。可以看出通過配位鍵交聯的有機硅彈性體具有很好的自修復能力,但是其力學強度仍有待提高,且酰氯材料價格昂貴,化學性質過于活潑需要低溫反應。
本文通過預聚體法,以2,6-二氨基吡啶(2,6-DAP)對聚二甲基硅氧烷預聚體進行擴鏈,將吡啶官能團引入主鏈中與Fe3+形成配位鍵,同時分子鏈中生成的酰胺基團之間能夠形成氫鍵作用,從而實現配位鍵和氫鍵協同作用,提高材料的拉伸強度(1.96 MPa),且保留較高的自修復效率(82.7%)。
1 實驗部分
1.1 原料和試劑
八甲基環四硅氧烷(D4):w=97%,道康寧公司;1,3-雙(3-氨基丙基)-1,1,3,3-四甲基二硅氧烷:w=97%,上海達瑞精細化學品有限公司;六亞甲基二異氰酸酯(HDI)、二苯甲烷-4,4'-二異氰酸酯(MDI)、二月桂酸二丁基錫(DBTL)、2,6-DAP:分析純,上海阿拉丁生化科技股份有限公司;四甲基氫氧化銨(Me)4NOH、六水合三氯化鐵、四氫呋喃(THF):分析純,阿達瑪斯試劑有限公司。2,6-DAP 使用前經甲苯重結晶,THF 使用前經過鈉絲回流除水,其他試劑使用前未經過純化。
1.2 測試與表征
傅里葉變換紅外光譜儀:美國熱電公司Nicolet 6700 型,掃描范圍4 000~650 cm–1,KBr 壓片涂膜及全反射;氫核磁共振譜儀:德國布魯克公司Advance 400 MHz 型,溶劑為氘代氯仿;紫外-可見分光光度計:日本島津公司UV-2550 型,將3.0 mg 樣品溶于10 mL THF 中配制稀溶液,取3 mL 溶液進行吸光度測試,波長范圍為250~600 nm;接觸角測量儀:上海中晨數字技術設備有限公司JC2000D2 型,以去離子水為測試介質;萬能拉力試驗機:美國SANS 美特斯公司E42.503 型,將樣品用裁刀裁成啞鈴型樣條,有效寬度4 mm,環境溫度25 ℃,拉伸速率為50 mm/min;激光共聚焦顯微鏡:日本基恩士公司VK-X100K 型;綜合熱分析儀:德國耐馳儀器制造有限公司STA409PC 型,氮氣氛圍,升溫速率10 ℃/min,測試范圍為室溫至900 ℃。
1.3 實驗步驟
1.3.1 端氨基聚二甲基硅氧烷(APT-PDMS)的合成 在250 mL 的三口燒瓶中,加入100 g D4和7.35 g 1,3-雙(3-氨基丙基)-1,1,3,3-四甲基二硅氧烷,以0.62 g (Me)4NOH 作為催化劑進行開環反應。反應在干燥的氮氣氛圍中進行,反應溫度為105 ℃。經過18 h 平衡反應后,將反應體系升溫至180 ℃以降解催化劑,隨后降溫至160 ℃,利用循環水泵抽真空(10 kPa),除去低沸點小分子雜質以及其他副產物,得到無色黏稠狀液體APTPDMS。合成反應方程式見圖1。
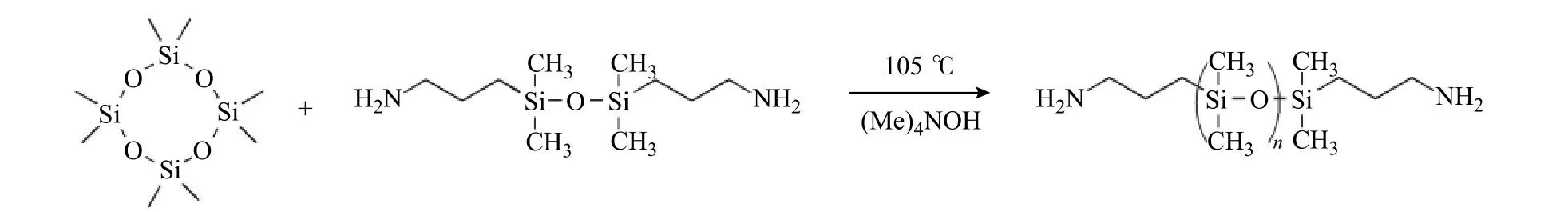
圖 1 APT-PDMS 的合成示意圖Fig. 1 Synthetic schematic diagram of APT-PDMS
1.3.2 自修復有機硅彈性體(PDMS-PU/M)的合成 首先,在100 mL 四口燒瓶中依次加入HDI(1.076 4 g)、MDI(1.067 6 g)、DBTL(0.05 mL)、THF(8 mL),置換氮氣,用鼓泡器隔絕空氣,待MDI 溶解均勻后,將體系逐漸升溫至60 ℃,利用恒壓滴液漏斗逐滴加入APT-PDMS 的THF 溶液(16 g/16 mL),在60 ℃下充分反應3 h,得到異氰酸酯封端的聚二甲基硅氧烷預聚體溶液。將0.58 g 2,6-DAP 溶解于10 mL THF 中,隨后將其緩慢滴加到預聚體溶液中進行擴鏈反應,繼續在60 ℃反應16 h 后,將溶液倒入培養皿中,在室溫環境下放置24 h 揮發溶劑,隨后在40 ℃的真空烘箱中干燥12 h,得到產物PDMS-PU。合成反應方程式見圖2。

圖 2 PDMS-PU 的合成示意圖Fig. 2 Synthetic schematic diagram of PDMS-PU
按照吡啶官能團和Fe3+的物質的量之比分別為5∶1、2∶1 和1∶1 將適量的六水合三氯化鐵加入PDMSPU 的THF 溶液(0.15 g/mL)中,依次記為PDMS-PU/M1、PDMS-PU/M2 和PDMS-PU/M3。溶液在50 ℃條件下充分攪拌1 h 得到均一的溶液體系,隨后將溶液倒入培養皿中,在室溫下放置24 h 揮發溶劑,接著在40 ℃下真空干燥12 h,得到PDMS-PU/M。
2 結果與討論
2.1 APT-PDMS 的結構表征
圖3 所示為APT-PDMS 的核磁共振氫譜,化學位移0.068~0.091 處的多重峰(a)為主鏈硅原子上所接甲基的質子峰,0.528 處的三重峰(b)為Si―CH2―CH2―CH2―NH2上亞甲基的質子峰,1.458 處的多重峰(c)為Si―CH2―CH2―CH2―NH2上亞甲基的質子峰,1.589 處的單峰(d)為–NH2上氨基的質子峰,2.665 處的三重峰(e)為Si―CH2―CH2―CH2―NH2上亞甲基的質子峰。對三重峰(e)與多重峰(a)進行積分,峰面積比為1∶69.33,計算得出APT-PDMS 的數均分子量大約為3.0×103,符合理論推算。在圖4 中所示的APT-PDMS 紅外光譜中,3 332 cm–1處是―NH2―的特征吸收峰,2 962 cm–1和2 905 cm–1處分別是主鏈硅原子上所接甲基的伸縮振動吸收峰,1 093 cm–1和1 020 cm–1處是主鏈Si―O 的伸縮振動峰。表明成功合成了APT-PDMS。
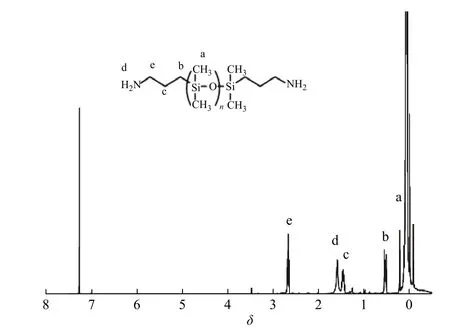
圖 3 APT-PDMS 的1H-NMR 譜圖Fig. 3 1H-NMR spectrum of APT-PDMS

圖 4 樣品的紅外吸收光譜Fig. 4 FT-IR spectra of samples
2.2 PDMS-PU 及PDMS-PU/M 的結構表征
2.2.1 PDMS-PU 及PDMS-PU/M 的紅外光譜表征 由PDMS-PU 的紅外譜圖(圖4)可知,在2 260 cm–1處無―NCO 的特征峰,對比APT-PDMS 的紅外譜圖,1 640 cm–1處酰胺鍵上羰基(C=O)的特征吸收峰及1 557 cm–1處―NH―的特征吸收峰的出現表明HDI 與MDI 已經完全反應,接入PDMS 分子鏈。1 599 cm–1處為MDI 上苯環的特征吸收峰。表明成功合成了PDMS-PU。PDMS-PU/M 與PDMS-PU 的紅外譜圖無明顯差別,其吡啶官能團與Fe3+之間的配位鍵作用由UV-Vis 進行表征。
2.2.2 PDMS-PU 及PDMS-PU/M 的UV-Vis 表 征
從圖5 中可以看出,PDMS-PU 中沒有發生配位的吡啶官能團在308 nm 存在明顯的吸收峰,當引入Fe3+后,在335 nm 出現了新的吸收峰,這是因為吡啶官能團及酰胺鍵與Fe3+之間生成了配位鍵,發生了電荷轉移,并且吸收峰強度隨著Fe3+與吡啶官能團比例的增大而增強,表明形成的配位鍵數目增多。此外,由于Fe3+配合物吸收低能量電磁波,導致在波長大于400 nm處有峰尾的存在,從而有力證明了PDMS-PU 與Fe3+之間形成了配位結構。
2.3 PDMS-PU/M 的力學性能表征

圖 5 PDMS-PU 和PDMS-PU/M 的UV-Vis 光譜Fig. 5 UV-Vis spectra of PDMS-PU and PDMS-PU/M
PDMS-PU/M 的力學性能分析如表1 所示,由表可知,隨著Fe3+與吡啶官能團物質的量之比增加,PDMS-PU/M 的拉伸強度從0.95 MPa 逐漸增加到1.96 MPa,但其斷裂伸長率從100.33%下降至73.15%。這是因為隨著Fe3+濃度的增加,PDMSPU/M 主鏈上吡啶官能團及酰胺鍵與Fe3+之間形成配位鍵的密度增大,使材料的拉伸強度有明顯提升,但限制了PDMS 分子鏈的運動,致使材料的斷裂伸長率略有下降。
2.4 PDMS-PU 的變溫紅外光譜表征
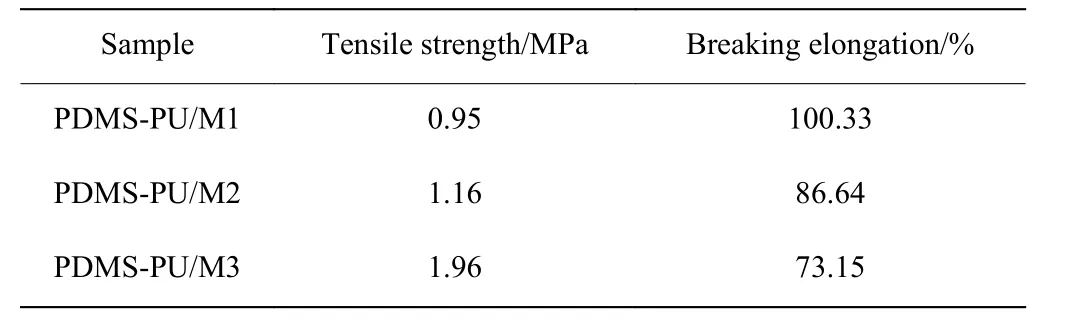
表 1 PDMS-PU/M 的力學性能Table 1 Mechanical properties of PDMS-PU/M
圖6 所示的變溫紅外吸收光譜探究了PDMSPU 分子鏈上氫鍵的相互作用。測試溫度為20~120 ℃,測試間隔為10 ℃。圖中的FT-IR 曲線顯示了形成氫鍵的酰胺鍵中C=O 及N―H 兩個特征峰的變化,1 640 cm–1處為酰胺鍵中C=O 伸縮振動吸收峰,1 557 cm–1處為N―H 的平面內彎曲振動吸收峰。圖6(a)為升溫過程,隨著溫度的升高,上述個特征峰分別向高波數和低波數移動,表明在升溫過程中,氫鍵遭到破壞。圖6(b)為降溫過程,最終兩個特征峰回歸原位,C=O 與N―H 重新形成氫鍵。由此可以看出,PDMS-PU 分子鏈間存在氫鍵的相互作用,且隨溫度的變化,氫鍵作用是可逆的。
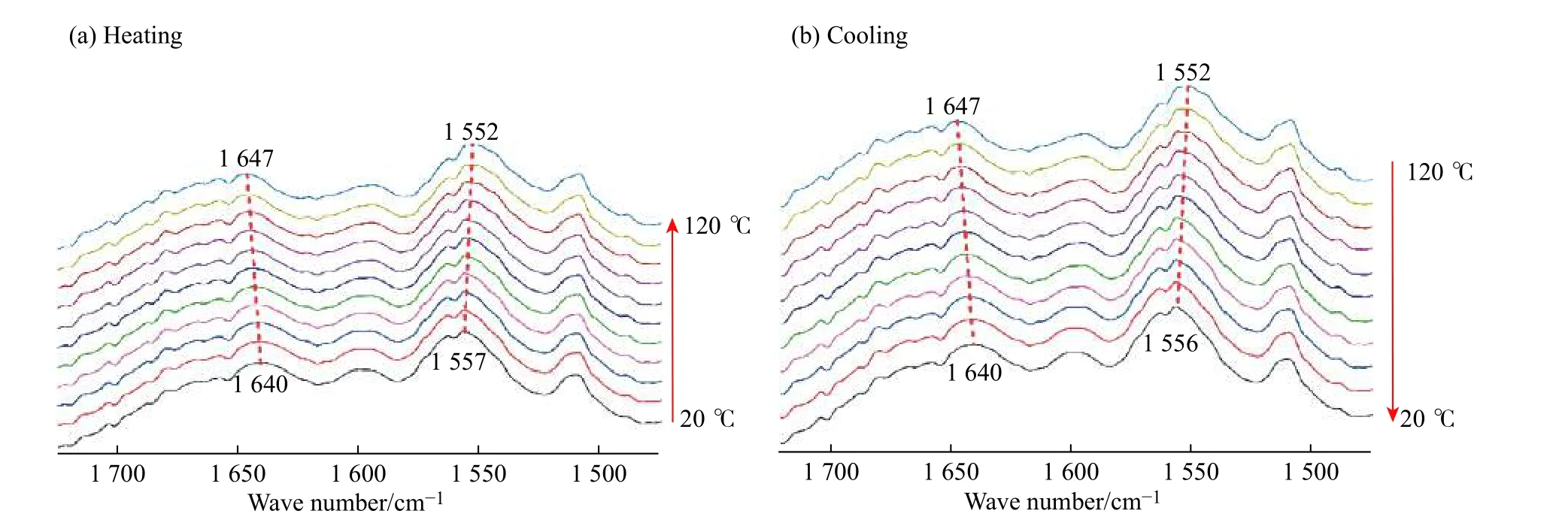
圖 6 PDMS-PU 的紅外吸收光譜Fig. 6 FT-IR spectra of PDMS-PU
2.5 PDMS-PU/M 自修復性能表征
2.5.1 PDMS-PU/M 自修復的形貌表征 有機硅彈性體中的配位鍵和氫鍵均為可逆非共價鍵,且硅氧烷主鏈具有優異的鏈運動能力,因此賦予了其優異的自修復性能。用美術刀將PDMS-PU/M2 膜切成兩部分,隨后立刻將這兩部分按照切口對齊拼在一起,在90 ℃條件下,PDMS-PU/M2 進行自修復(圖7),每隔1.5 h 取樣,以激光共聚焦顯微鏡對其進行形貌表征。從圖中可以看出,修復過程中裂縫邊緣的材料呈現出微觀流動狀態,在重力和表面能的驅動下,分子鏈向缺陷處運動,以減少材料表面積,降低材料整體表面能,實現修復,整個自修復過程在6 h 后完成,寬60 μm、深244.7 μm 的裂縫基本消失。

圖 7 PDMS-PU/M2 在90 ℃下的自修復過程Fig. 7 Healing process of PDMS-PU/M2 at 90 ℃
2.5.2 PDMS-PU/M 自修復的力學性能表征 用美術刀將PDMS-PU/M 膜切成兩部分,隨后立刻將這兩部分按照切口對齊拼在一起,分別放置于40、60 ℃和90 ℃的烘箱中6 h 進行自修復。修復后,測得的應力-應變曲線如圖8 所示。自修復效率(EH)計算公式為,EH=(σhealed/σoriginal)×100%,其中σhealed代表修復后材料的拉伸強度,σoriginal代表原始材料的拉伸強度。在圖8 中,隨著自修復溫度的升高,分子鏈的運動能力增強,材料自修復能力和效率得到提高。
表2 為PDMS-PU/M 自修復后的力學性能,由表2 可以看出,隨著Fe3+在PDMS-PU/M 中所占比例提高,材料的自修復效率呈現先增大后下降趨勢,這是因為隨著Fe3+濃度增大,分子鏈中吡啶官能團及酰胺鍵與Fe3+形成的配位鍵數量增多,動態交聯密度增大,受外力斷開的分子鏈有更大的幾率重新與Fe3+形成新配位鍵,實現修復,從而提高材料的自修復效率。但隨后因為交聯密度繼續增大,分子鏈間和分子鏈內的配位鍵作用力過強,阻礙了分子鏈運動,在修復過程中,分子鏈中的吡啶官能團及酰胺鍵難以與Fe3+接觸,形成新配位鍵,所以自修復效率呈現下降趨勢。但隨著Fe3+濃度增加,在相同條件下修復后的材料其拉伸強度均有所提高。樣品PDMS-PU/M3 自修復后拉伸強度達到最大值(1.62 MPa)且具有較高自修復效率(82.7%)。值得注意的是PDMS-PU/M2(90 ℃)的自修復效率為104.3%,力學性能優于原始材料,其原因是樣品以溶劑揮發成膜工藝制備,在溶劑揮發過程中因表面溶劑揮發速率快,其膜內部仍有微量溶劑未能及時揮發,隨后在真空干燥過程中被去除,因此留下微孔,導致材料表面及內部存在部分缺陷使其力學性能下降,在90 ℃自修復的過程中,其分子鏈之間氫鍵和配位鍵的相互作用減弱,分子鏈活動能力得到增強,在一定的時間條件下,除了實現對材料受損切面的修復,還能夠對材料原有缺陷進行修復。
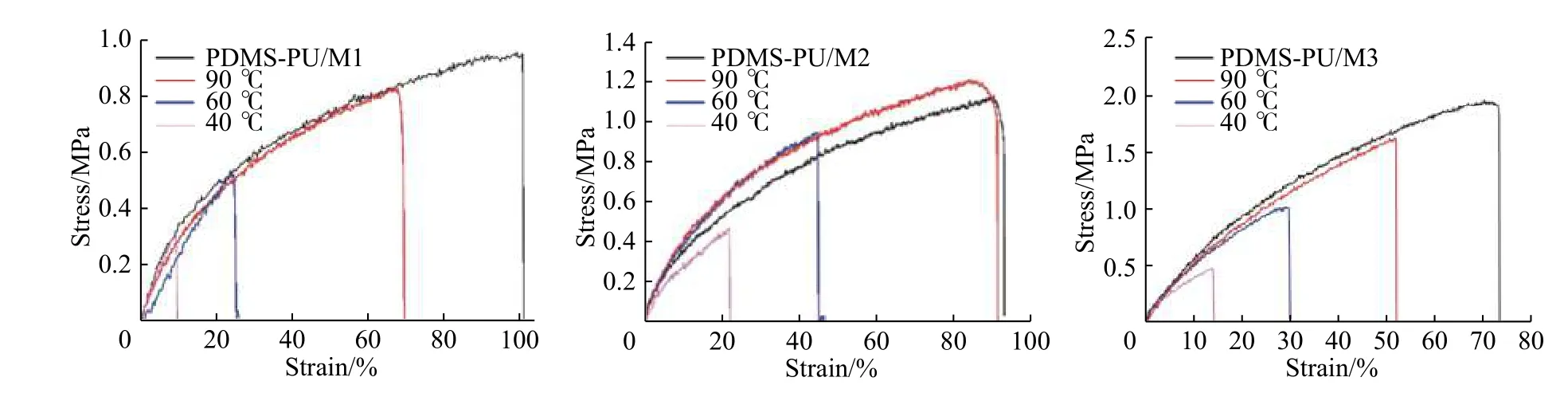
圖 8 PDMS-PU/M 原始材料及在不同溫度下修復6 h 后的應力-應變曲線Fig. 8 Stress-strain curves of original and healed PDMS-PU/M for 6 h at different temperatures

表 2 PDMS-PU/M 自修復后的力學性能Table 2 Mechanical properties of PDMS-PU/M after self-healing
2.6 PDMS-PU/M 的熱穩定性表征
圖9 為PDMS-PU/M 的熱重分析曲線,分析表明樣品均具有優良的熱穩定性,在修復溫度范圍內未發生降解,失重5%的溫度約為300 ℃,最大降解溫度約為500 ℃。
3 結 論
(1)以D4為原料,通過開環反應成功合成了端氨基聚二甲基硅氧烷。
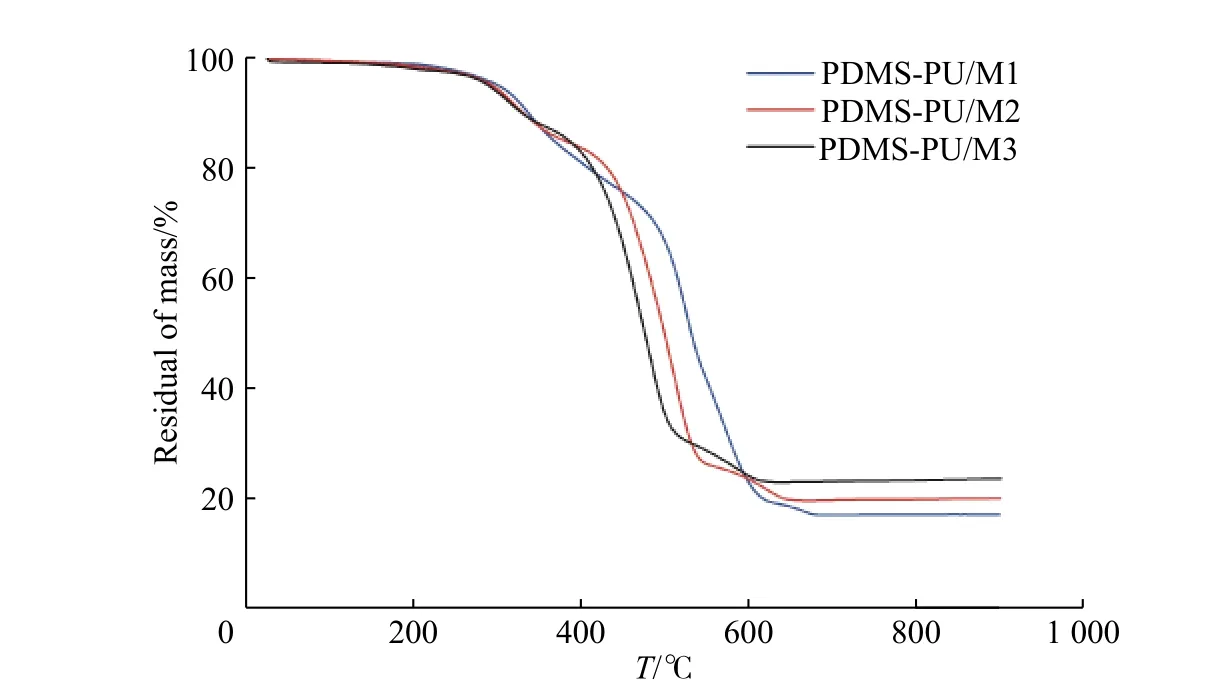
圖 9 PDMS-PU/M 的TG 曲線Fig. 9 TG curves of PDMS-PU/M
(2)通過預聚體法,以二氨基吡啶為擴鏈劑合成了PDMS-PU 彈性體,用Fe3+配位交聯,制備了自修復有機硅彈性體PDMS-PU/M,其具有良好的疏水性能、優異的力學性能和出色的自修復效率。當吡啶官能團與Fe3+的物質的量之比為1∶1 時,其拉伸強度可達1.96 MPa,自修復效率為82.7%。
(3)隨著Fe3+濃度的增加,材料的自修復效率呈現先升后降的趨勢,但其力學性能均得到增強。隨溫度的升高,自修復效率提升。
猜你喜歡
材料與冶金學報(2022年2期)2022-08-10 09:15:46
云南化工(2021年11期)2022-01-12 06:06:14
山東冶金(2019年3期)2019-07-10 00:54:00
中國鑄造裝備與技術(2017年3期)2017-06-21 11:33:46
中國塑料(2016年6期)2016-06-27 06:34:16
西安工程大學學報(2016年2期)2016-06-05 12:25:17
中國塑料(2015年12期)2015-10-16 00:57:14
中國塑料(2015年9期)2015-10-14 01:12:26
中國塑料(2015年4期)2015-10-14 01:09:18
焊接(2015年9期)2015-07-18 11:03:53
