固態電解質鋰鑭鋯氧(LLZO)的研究進展
2020-04-04 05:21:52姜鵬峰石元盛李康萬韓百川顏立全
儲能科學與技術 2020年2期
關鍵詞:界面
姜鵬峰,石元盛,李康萬,韓百川,顏立全,孫 洋,盧 俠
(1中山大學,廣東 廣州 510006;2北京化工大學,北京 100029)
鋰離子電池(LIBs)已廣泛應用于各類便攜式電子設備中。新能源汽車的興起更是促進了LIB技術的顯著進步。考慮到目前新能源汽車的里程焦慮及未來我國巨大的儲能需求,發展下一代高能量密度,高安全性的電池具有重大意義。目前,有機系液態電解質體系在商用中取得了巨大的成功,但在更高能量密度、可靠性和安全性的應用方面,現有的LIBs還是顯示出了一些局限性:使用有機電解液,特別是含有環狀碳酸酯、鏈狀碳酸酯、羧酸酯類電解液具有高度易燃性[1];電解液具有較強的腐蝕性且容易分解生成CO、H2S、LEL等氣相產物[2];傳統的有機電解液與高電壓(≥5 V)正極及大容量金屬鋰負極不相匹配[3]。在過去的幾十年里,如何解決鋰電池存在的漏液及自燃問題也一直是廣受關注的焦點。解決上述問題選擇之一是使用無機固態電解質。固態電解質不僅能夠替代電解液,而且為新型Li-O2、Li-S電池的開發提供了可能性[4]。使用固態電解質的電池體系安全性高、無漏液現象,且固態電解質也可作為電池隔膜使用,簡化了電池的整體設計。固態電解質可能比有機電解液普遍具有更寬的電化學窗口,有利于進一步拓寬電池的電壓范圍,提升能量密度。固態電解質還可以支持電池在高低溫(例如200oC到-50oC范圍內)下工作。使用固態電解質總體提高了電池的安全性能和使用壽命。
無機固態電解質由晶體鋰離子導體和非晶體鋰離子導體兩類組成。晶體鋰離子導體又分為:鋰超離子導體(LISICON)結構(如Li1+xZn1-xGeO4)[5]及其硫代(thio-LISICON)結構(如Li4-xGe1-xPxS4)[6]、鈉超離子導體(NASICON)結構[Li1+xMxTi2-x(PO4)3,M3+:Al3+、Y3+、In3+…][7]、石榴石結構[Li7La3Zr2O12(LLZO)][8]、鈣鈦礦結構[Li3xLa2/3-x□1/3-2xTiO3(LLTO),□=空位][9]和反鈣鈦礦結構(Li3OX,X=Cl、Br)[10]、鋰的氫化物 [如 Li2(NH)、Li(BH4)][11]、鋰的鹵素化合物(Li2MCl4,M:Mg、Mn、Fe、Cd)[12]、硫銀鍺結構(Li6PS5X,X:Cl、Br、I)[13]。而非晶體鋰離子導體包括:鋰的硫化物玻璃陶瓷(Li2S-P2S5[14]和Li2S-SiS2[15])、鋰的氮化物(Li3N[16]和LiPON[17])等。這些固態材料作為電解質用于全固態鋰離子電池的構筑有其各自的優點和缺點[18-21]。在諸多固態電解質中,石榴石型固態電解質自從被發現以來便被廣泛研究。石榴石型固態電解質具有以下優越的物理和化學性質[19,22-25]:室溫下具有高離子電導;對金屬鋰具有良好的化學穩定性;寬的電化學穩定窗口。這些突出的優點無疑使得LLZO成為理想的用于構筑全固態電池的固態電解質之一。與其他鋰含量較低的石榴石相比,通過采取適當的摻雜策略和優化燒結工藝,LLZO可以實現更高的電導率(室溫下約1 mS/cm)。研究人員也一直在探索可實現商業化的LLZO用于構筑固態電池的工藝設計路線。對于實際的固體器件來說,使用固態電解質構筑電池所面臨的主要挑戰包括與金屬負極(Li、Na、K)匹配、界面間良好物理接觸及界面穩定性,而解決這些問題的關鍵在于透徹地理解固態電解質材料的基本特性。
本文總結了近年來富鋰石榴石的研究成果,對LLZO的結構、Li離子輸運機制及構筑石榴石型固態電池工藝技術等方面進行了梳理。
1 石榴石型固態電解質的結構
石榴石結構的化學通式為:A3B2(XO4)3(A=Ca、Mg、Fe、Mn;B=Al、Fe、Cr、Ti、Zr、V),其中,A、B、X分別有8、6、4個氧配位[26]。當X為Li+時,其具有Li+導通能力。Li(X)原子處于框架結構的空隙中。石榴石晶體結構為面心立方(FCC),空間群為Ia-3d。通常,按每結構單元含有的Li+的數量將含鋰的石榴石型固態電解質分為Li3、Li5、Li6以及Li7體系。1969年,Kasper等[27]制備了Li3體系石榴石型電解質,在該結構中,Li鋰離子全部位于間隙空間最小的四面體位(24d)中,八面體位置沒有Li占據。由于Li—O鍵較強,Li-Li間距遠,Li離子被束縛在四面體中難以遷移,所以Li3體系電解質離子電導率較低。2003年,Weppner等[28]首次對Li5體系石榴石型快離子導體進行報道,其室溫離子電導率為10-6S/cm數量級。相對于Li3來說,Li5體系多出2個Li,5個Li將重新排列,導致晶體結構中80%的四面體被占據,40%的八面體被占據。位于八面體的Li是可移動的,并且同時引入了四面體的空位,為Li的移動提供了更多潛在的跳躍位點。Li位置的多樣性和部分占據的位置使得Li5體系比Li3體系具有更高的離子電導率。但是因為Li2(16f)位置Li離子的濃度不高,所以電導率僅有很小的提升。通過二價堿土離子取代La3+,可以進一步提高Li+濃度,得到通式為Li6La2M2O12的Li6體系。摻雜替代La(A位,24c)獲得的Li6BaLa2Ta2O12的室溫電導率可達4×10-5~5×10-5S·cm-1,活化能為0.4 eV[28-29]。然而堿土金屬離子半徑一般小于La,導致摻入La位后晶胞尺寸縮小,Li—O鍵長變短,Li離子不易遷移,電導率與Li5體系相比提升不大。
提高電導率的關鍵在于制造四面體空隙24d位置的鋰空位。從Li3到Li5、Li6體系,增加單胞中的Li+的數目可以讓四面體空隙位置的Li+減少,八面體空隙位置的Li+增加,進而提高電導率。2007年Murugan等[8]首次采用固相合成法,在1230oC下燒結制備出了純立方相石榴石型結構[a=12.9682(6)?;空間群Ia-3d]的Li7La3Zr2O12(LLZO)。隨后,Kaeriyama等[30]于1180oC燒結同樣合成出純立方相LLZO。LLZO存在立方相(c-LLZO)和四方相(t-LLZO)兩種晶體結構,兩種晶體結構示意圖及Li1、Li2配位多面體如圖1(a)、(b)和(d)所示。兩種結構最顯著的差別就是Li的占位,在立方相中Li部分占據間隙位,而在四方相中Li占滿間隙位。t-LLZO的離子電導率比c-LLZO低了兩個數量級,約在10-6S/cm數量級。t-LLZO與c-LLZO存在空間群差異及離子電導率差異,具體見表1。

表1 四方、立方LLZO的基本性質Table 1 Structural fundamentals of t/c LLZO
基于諸多純相LLZO的研究發現[8,34-37],其晶格常數通常在a=12.95~12.97 ?范圍之間。石榴石型LLZO的晶體結構由8配位的LaO8十二面體(24c)和6配位的ZrO6八面體(16a)構成。Li原子在兩種位點內隨機占位且部分占據框架結構的間隙,即四面體24d位點和八面體48g或是遠離中心的96h位點。通常情況下,由于Li+共面時存在排斥力,48g位點取代了96h位點。相比于Li3體系與Li5體系的立方相結構,Li7體系中四面體位置的Li+占有率有所降低,而八面體位置的Li+占有率有所提高。
對比t-LLZO,如圖1(f)所示,c-LLZO的Li1占據率接近1。對于c-LLZO,Li1位置作為連接點,其Li-Li的距離較小,而t-LLZO,Li-Li的距離大于2.5 ?。躍遷距離的縮短是c-LLZO具有高離子電導率的原因之一。t-LLZO中四面體空隙有兩種位置,分別是8a和16e位;八面體空隙同樣有兩種位置,分別是16f和32g位,如圖1(c)所示。其中Li1原子占據四面體8a位置,另一種四面體位置沒有占據,Li2和Li3完全占據扭曲的八面體位16f與32g位。根據晶格內Li離子互斥模型[38],Li1僅僅占據1/3四面體空隙8a位,余下的占據2/3四面體16e位。Li2占據全部的八面體空隙16f和32g位。對比t-LLZO與c-LLZO的晶胞參數可知,t-LLZO中8a與16e位間距較遠,因此允許在8a和32g位上容納兩個Li離子。而32g與16f位間距較近,使得毗鄰32g位的16e位無法容納Li離子。t-LLZO中3個鋰位置均為全充滿狀態且鋰離子間距較大,造成Li離子遷移困難,離子電導率較低。c-LLZO可容納鋰的位置同樣有3種,分別是四面體空隙24d位、正八面體空隙48g位和偏八面體空隙96h位。由于Li離子間存在斥力,Li離子一般占據八面體中心48g位的兩側,即96h位。96h位距離很近,僅一半可被Li離子占據。c-LLZO中鋰計量數小于7,表明Li離子在24d和96h位上處于隨機無序占據狀態[39]。Li離子在c-LLZO中的遷移容易很多,故而該材料表現出高電導率。環繞八面體的鋰離子通道示意圖如圖1(e)、(f)所示。不同于t-LLZO[圖1(e)],L1與L2位為相間排列。因為Li+間的排斥作用,鋰離子以無序方式相隔占據L1、L2位。此外,基于第一性原理計算發現,四方相中Li離子是集體移動的,而立方相的LLZO中Li離子是非共時的單離子躍遷和誘導下的集體輸運,相比前者,后者離子躍遷的能壘更低。
在較高的溫度下,四方相可以向立方相轉變。實驗觀察到的相變溫度是變化的。室溫下,c-LLZO是一種鋰離子無序的亞穩定相。隨著晶格中的Li離子增加到24d和96h位容納的上限,電荷排斥力的存在會讓Li離子穩定到四方相的格位上,物相也隨之轉變為四方相。Adams等[40]基于原位X射線衍射(In-situ XRD)結果報道了純相LLZO在177oC處發生四方相向立方相的相轉變過程,而Geiger等[41]使用XRD表征這種相轉變發生在100~150oC之間。Kokal等[42]使用差示掃描量熱法(DSC)證明,相變溫度遠高于700oC。設法在室溫下穩定立方相,以利用它的高離子導電性是目前科研人員關注的重點。大量的工作集中在摻雜元素的引入及使用各種合成方法以穩定立方相。這些合成方法通常需要在750~1230oC范圍內進行多次煅燒,總合成時間接近或超過24 h,具體的內容將在下一章詳細展開。
諸多實驗研究了燒結溫度對LLZO晶體結構的影響[8,39-40,42-45]。研究結果表明LLZO在1150~1230oC的溫度范圍內形成穩定的立方結構,而在1150oC的燒結溫度以下,傾向形成四方結構。2009年Awaka等[46]報道了在較低溫度980oC燒結后LLZO存在四方結構(空間群I41/acd)。Percival等[47]也報道了在750oC以下燒結得到的石榴石型Li7La3Sn2O12具有四方結構。研究結果表明,在室溫條件下,t-LLZO是穩定的結構,t-LLZO是一種微變形的立方石榴石型結構[46]。其c軸晶胞參數變小,而a、b兩軸晶胞參數變大。如表1所示,文獻報道的t-LLZO晶格參數為a=13.07~13.12 ?,c=12.67~12.72 ?[48-51]。在立方相向四方相轉變(四方畸變)時,立方相24d位轉化為完全占據的8a位和未占據的16e位,立方相96h位轉化為兩個16f和32g位。
t-LLZO較c-LLZO的室溫離子電導率低兩個數量級。Wolfenstine等[48]采用熱壓法制備了t-LLZO樣品。其電導率在室溫下為2.3×10-4S/cm,是報道的t-LLZO所具有的最高離子電導率。值得注意的是,熱壓得到的樣品具有高達98%的相對密度,可能是熱壓t-LLZO樣品具有高電導率的主要原因。綜上所述,c-LLZO比t-LLZO具有更高的電導率。對于立方相結構,隨著總的Li濃度的提高,燒結溫度的升高,八面體位置的Li濃度增加,石榴石的體相離子電導率增大。
2 Li+在LLZO中的輸運
盡管c-LLZO具有較高的離子電導率,但對比傳統液態電解質還有一定差距。清晰地理解Li+在石榴石結構中的輸運,有利于進一步提高該材料的離子電導率。得到鋰離子的具體占位是分析鋰離子在晶格中輸運機制的第一步。Awaka等[52]通過對c-LLZO的單晶X射線衍射數據精修發現,四面體配位的Li1(24d)位更容易被占據。但由于Li+的XRD散射因子很弱,使用粉末X射線衍射的數據定位Li5,Li7石榴石中Li+的占位存在很大的爭議。中子衍射可以很好的解決這個問題。2011年,Goodenough等[53]得到從室溫到600oC下立方相的Li7La3Zr2O12中子衍射數據(圖2)。通過最大熵的分析方法可清楚的發現,位于24d與96h位的鋰表現出明顯的離域,而當溫度高于400oC,不同占位的Li離子由不連續核密度分布,逐漸互相連通構成24d→96h→48g→96h→24d的3D輸運通道。此外隨著溫度升高,位于扭曲八面體中心的48g位的Li占據率逐漸上升。
核磁共振(NMR)可以探測固體中短程內的結構變化,很適合分析Li的配位環境[54]。24d與96h位的Li離子的化學環境存在略微的不同,高分辨率的6Li-NMR可將二者區分開來。進一步通過二維核磁共振法則可以探測到這兩種位置的Li離子之間的交換過程。在Li3體系中(如Li3Nd3TeO12),6Li NMR測試結果只得到了一種鋰的信號峰,該峰對應著四面體位置上Li的有序排列[55]。而對于Li5、Li6和Li7體系,Li NMR研究結果得出了第二種信號峰,該信號峰對應扭曲的八面體(48g/96h)位上的Li。對于富鋰體系,四面體位置的Li離子沒有參與傳導,而偏八面體位置的Li具有較高的移動性。Wullen等[56]同樣利用NMR對Li5體系(Li5La3Nb2O12)進行了研究。研究發現八面體位置的Li占有率增大,其離子電導率增大,這說明八面體位置的Li可能具有更高的移動性。上述現象可能是Li5體系具有較高離子電導率的主要原因。此外,在60~140oC的溫度范圍內,四面體位置的Li所對應峰的寬度沒有發生變化,表明四面體位置的Li的移動性較低。二維交換NMR測試結果表明,Li八面體位置之間存在快速的交換過程,但在四面體與八面體位置之間,沒有Li交換過程,因此認為Li的躍遷只發生在相鄰八面體之間,而不經過四面體位置。Wang等[57]使用梯度核磁共振技術清晰觀察到了24d與96h之間的離子交換,并發現鋰沿著24d→96h→48g→96h→24d路徑進行輸運,八面體與四面體占位的離子的擴散系數分別為10-9cm2/s和10-11cm2/s。測試結果如圖3所示。
近年來,先進的實驗技術和表征手段有效地幫助理解石榴石結構中鋰離子的輸運過程。但是在原子尺度上皮秒量級的離子躍遷,目前實驗表征技術仍難給出清晰的答案。2012年Xu等[58]使用第一性原理細致計算了Li3、Li5、Li7石榴石體系的Li離子占據態與擴散過程。結果顯示持續增加晶格中Li的濃度會削弱Li1(24d)位的穩定性。通過對c-LLZO結構優化發現,在基態能量下的構型中,超過一半的四面體Li1(24d)位是沒有鋰離子占據的,而八面體的Li2位占據率增大到90%。后續過渡態計算發現,石榴石結構中Li的輸運路徑有兩種[圖4(a)、(b)、(c)]。在路徑A中,鋰離子通過相鄰的八面體位點之間的間隙遷移,繞過與之連接的四面體。在路徑B中,鋰離子通過八面體和四面體的共用三角形面進行躍遷。路徑A中的鋰離子遷移的活化能為0.8 eV,當材料Li+含量較低時(例如Li5La3Nb2O12),鋰離子遷移路線傾向于路徑A;路線B中的鋰離子遷移的活化能為0.26 eV,在高鋰填充物(例如Li7La3Ta2O12)中更傾向于路徑B。上述工作指出優化Li/Li空位的比率,使以路徑B擴散過程占據主導是非常重要的。但該工作沒有考慮LLZO晶格中存在著大量Li/Li空位排列的動態過程。次年,Jalem等[59]使用第一性原理分子動力學模擬方法,發現LLZO中高體相電導率是由于協同擴散主導的,在24d位占據的Li是不穩定的,其可能誘導臨近的Li發生再分布。He等[60]分析快離子導體(LGPS、LLZO、LATP)AIMD模擬結果發現[圖4(d)、(e)、(f)],Li+動力學的 Van Hove關聯函數顯示在幾個皮秒量級下大多數Li+是以高度協同的方式進行躍遷的。在LLZO中,四面體的鋰離子躍遷到最近鄰O的位置并占據這些O的位置,然后再躍遷到最近鄰的四面體位置,最終展現為沿石榴石擴散通道下的多離子躍遷的過程。
除了上述討論的微觀擴散路徑與機制,通常認為晶界對離子的輸運有阻礙作用。晶界為多晶樣品中不同取向的晶體之間的接觸表面,它們在結構和組成上與體相晶體有很大的不同[61]。晶界已被證明在大多數情況下增加了離子跨越體相遷移的阻力,使它們在宏觀樣品中的傳輸過程變得不穩定[61]。正空間電荷(陰離子空位)可以使移動陽離子的晶界發生排斥作用且某些材料的晶界也可以通過形成不協調位點的通道來輔助平行于其表面的離子傳導過程[62]。Yu等[63]的計算結果表明,Li離子在LLZO晶界區域的傳輸通常比在體相區域要慢。值得注意的是,此過程對晶界結構及溫度十分敏感。鋰離子在高溫(>900 K)下,在LLZO晶界中的擴散僅比體相擴散稍慢一點。在室溫下,鋰離子在相對緊湊的晶界處離子電導率為體相的一半,而在相對松弛的晶界處則比體相的離子電導低兩個數量級。Dobretsov等[64]的實驗結果表明,LLZO樣品晶界電阻明顯大于體相電阻,這種電阻的增加很可能是由位于晶界上的未知成分所決定的,而不是LLZO本身。通過XRD及活化能的計算結果,存在于LLZO晶界上的雜質相可能為LiZrO2或LiAlO2相。一般來說,調控晶粒晶界的濃度可以改變固態電解質的離子電導率,這對提高LLZO的離子電導率具有一定的指導意義。
3 元素摻雜對LLZO晶體結構和離子電導率的影響
目前,摻雜離子半徑與主離子相近的元素是促進c-LLZO在室溫下穩定和提高離子電導率的策略選擇。通過置換或者摻雜離子(即與主離子價態不同的摻雜劑)可以產生電荷補償的空位或間隙。陽離子取代在維持氧化學平衡的同時通過降低Li含量或增加Li空位濃度來穩定立方相。穩定c-LLZO所需的鋰空位的確切數量尚未確定,通常認為此數值處于每單位0.125~0.500 mol的鋰空位之間[65-68]。具有較高Li空位濃度(0.4~0.5 mol)的LLZO在室溫下的離子電導率同樣較高。此外,共摻雜技術也得到了廣泛的應用。共摻雜不僅可以穩定立方結構,而且可以有效促進Li的輸運。此外,密度泛函理論(DFT)計算了LLZO中摻雜物可能的位置,并為尋找具有優異性能的LLZO基復合材料提供選擇[69],已經進行的取代包括:Al、Fe、Ge和Ga來取代 Li; Sr,Y 和 Ce 取代 La;Nb、Ti、Ta、Sb、Mg、Sc、Zn、Ru、W、Te取代Zr等。表2列出了未摻雜和摻雜LLZO的燒結溫度、燒結時間、晶格常數、相對密度和離子傳導活化能的詳細信息。
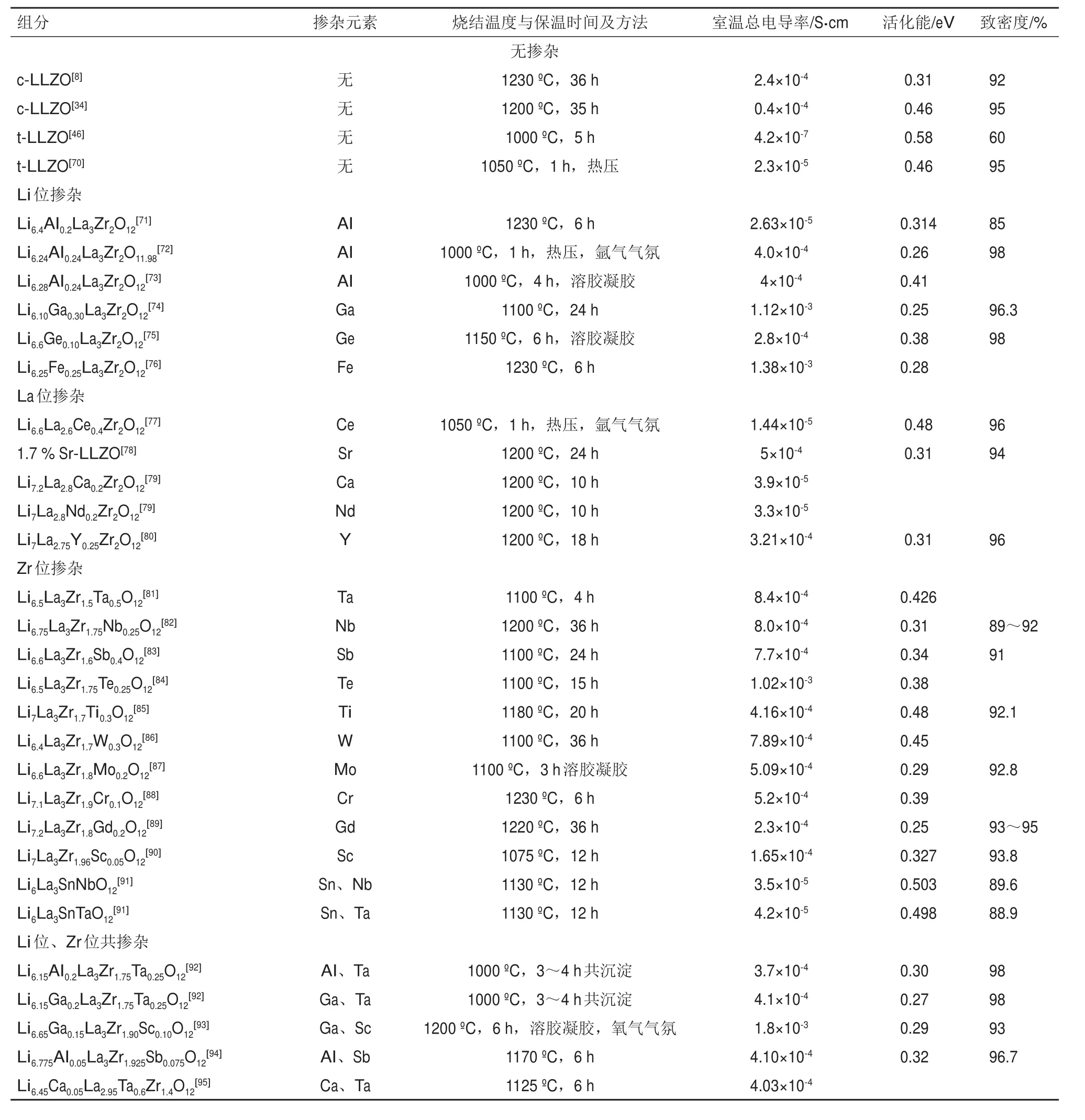
表2 LLZO的摻雜組分、燒結條件、總電導率、活化能及致密度Table 2 Doping compositions,sintering conditions,total conductivity,activation energy and density of LLZO
c-LLZO中可摻雜位置共有3種:分別是Li位、La位、Zr位。其中關于Li位和Zr位的摻雜研究最為廣泛。總結發現,無論LLZO中鋰含量是恒定的還是變化的,無論被取代的位置是什么,也無論相應的取代元素是什么,當晶格參數在12.91~12.98 ?范圍內時,鋰離子電導率最高。晶格參數超出此范圍的取代則表現出較低的離子電導率。Kihira等[105]已經系統地研究了LLZO最佳晶格參數,在該研究中用Nb代替Zr,用各種離子尺寸的堿土金屬(Mg、Ca、Sr和Ba)代替La,實驗結果表明:不論是Li含量固定的組分還是因元素摻雜Li含量發生變化的組分,最佳晶格常數均在12.94~12.96 ?之間。在石榴石系列的所有其他成分中觀察到的趨勢同樣表明,具有高離子電導率的LLZO的最佳晶格參數也都處于此范圍內。Zeier指出[106],對于石榴石型晶體結構來說,最佳的晶格常數范圍是12.90~12.95 ?。擴展的晶格能夠在一定程度上提高離子導電性。目前的工作尚未嚴格討論在晶格參數范圍內存在最大鋰離子導電性的機理。一項計算研究指出了關于內部結構的具體參數,諸如八面體和四面體Li位點的大小或體積以及三角形窗口是如何隨摻雜物變化而變化的[107]。在摻Ta的LLZO中存在晶格收縮現象,鋰離子的含量和表面積都增加,空位同樣增加。與Zr相比,Ta對鄰近的O離子的吸引力更強,由于Ta—O鍵收縮,LaO8的8個扭曲的立方結構則保持剛性,最終增加了位點大小和頸部面積。
一般認為,摻雜劑(如Al3+;燒結過程從坩堝中部分取代LLZO或直接摻雜)可以穩定立方相。Geiger等[108]研究了Al摻雜和燒結溫度對Li7La3Zr2O12結構的影響。Al存在于LLZO晶體結構中的兩個Li位置上。在沒有Al摻雜和較低燒結溫度下得到LLZO為四方相結構。Kotobuki等[109]報道了Al2O3作為燒結助劑可以降低c-LLZO的燒結溫度(約200oC),并且抑制La2Zr2O7雜質的生成。對于摻雜Al的化合物,晶格會隨著填滿四面體位置的鋁離子的增加而擴展。隨著Al摻雜量的增加,位點大小沒有明顯變化,但頸部尺寸增大。研究人員通過中子衍射、27Al-NMR測試手段研究了Al在c-LLZO晶格中的位置[109-111]。結果為四面體空隙24d位和八面體空隙96h位與Li+競爭。一個Al3+可以向晶格中引入3個Li空位,同時Al3+在四面體中的遷移也會相對Li離子困難得多。盡管Al3+占據了四面體的位置,對Li的傳導有一定阻礙作用。但因為增加了平均頸部尺寸,所以Al的引入實際是有利于提升離子電導率的。值得注意的是,不同元素摻雜對LLZO離子電導率的提升存在一定濃度范圍。在此濃度范圍之外,摻雜劑的濃度要么低到無法對結構產生影響,要么高到無法被容納在石榴石結構框架中。元素摻雜同樣可能導致Li濃度升高或降低。過度偏離的晶體結構與Li濃度都將導致Li離子電導率下降。最高導電率的LLZO基化合物的Li含量為每單元6.1~6.8,低于每單元7.5的理論極限。Zeier已經指出了石榴石族的最佳極限,迄今為止已實現最高電導率的為Li6.55Ga0.15La3Zr2O12,室溫下可達2.06×10-3S/cm[106]。Jalem等[112]使用分子動力學模擬計算得出,Li7-3xGaxLa3Zr2O12(x=0.02)離子電導率可以達到6.08×10-3S/cm。該值很可能是石榴石型固態電解質鋰離子電導率的上限。迄今為止,LLZO離子電導率的最高值仍比普通電解液低一個數量級。
4 構筑LLZO基固態電池的嘗試及展望
當前高能量密度鋰電體系仍依賴于高鎳三元正極、硅碳負極和電解液的組合。商業化的鋰電池有望在未來5年內達到350 W·h/kg的極限,但這仍無法滿足動力電池在能量密度方面日益增長的要求。因此全固態電池被推到了前端,構筑LLZO基固態電池在最為關鍵的能量密度方面,有望徹底解決純電動汽車的里程焦慮。目前商用體系下的鋰離子電池已經逐步接近極限性能,特斯拉NCA 18650電芯下的電池組,能量密度為250 W·h/kg,應用于Model 3的21700電芯能量密度更是高達300 W·h/kg,支持續航里程400~500 km。如此高的性能也仍然無法解決續航里程焦慮。根據《汽車產業中長期發展計劃》《節能與新能源汽車技術路線圖》的相關指引,動力電池系統的能量密度需要在2025—2030年內達到350 W·h/kg以滿足市面上電動汽車的續航里程需求。從目前技術來看,僅憑借傳統鋰電池的技術研發,這一目標顯然已經無法實現。而為保證動力電池的高能量密度和安全性,固態電池的研發進度給整個新能源汽車行業帶來了光明。因此,全固態電池被廣泛認為是下一代動力電池正確的技術研發方向。不過固態電池雖然具有諸多優勢,但根據現有的研發進展來看,也還有兩項技術難題尚未攻破。一是固態電解質在室溫條件下的離子電導率不高,二是固態電解質與正負極之間界面阻抗比較大。
絕大多數基于氧化物和硫化物的固態電解質材料在與鋰金屬接觸時是熱力學不穩定的,這可能導致在二者界面處形成具有不同性質的新相或混合物。這種界面一般具有較差的離子導電性,導致高的界面阻抗甚至是成分降解。固態電解質用于構筑全固態鋰電池的一個潛在缺點是離子擴散依賴于固體顆粒間的接觸。低離子電導率與高界面阻抗導致了固態電池的高內阻,鋰離子在電池內部傳輸效率低,在高倍率大電流下的運動能力更差,直接影響電池的能量密度與功率密度。在多晶材料和復合電極中,為了實現高效離子傳導,必須最大限度地保持固體顆粒之間的接觸。發展有效的策略來解決物理接觸的問題是構筑固態電池的當務之急。LLZO與金屬Li有著優異的電化學穩定性,但許多研究報道了其具有較高的面積比阻抗(ASR)。典型的阻抗值達到數百個kΩ·cm2,這些研究表明Li與LLZO粘附性差或LLZO表面雜質(如Li2CO3)導致了Li和LLZO之間較差的物理接觸。為了改善金屬Li與LLZO界面接觸差的問題,當前采用較多的技術手段包括原子層沉積(ALD)、濺射、濕法處理、熱蒸發、拋光、加熱等[113-119]。在已報道的技術中,將Li加熱熔化以改善界面接觸是最直接且簡單的方法。據報道[120-121],在加熱后,界面電阻在室溫下下降至 25~28 Ω·cm2。Li2CO3鈍化層的形成同樣會阻礙Li對LLZO表面的粘附。Sakamoto等[122]已通過試驗證明在LLZO表面加熱金屬Li之前,采用濕法(乙二醇基添加劑)拋光石榴石表面的策略,可以更進一步降低Li-LLZO界面電阻至2 Ω·cm2。Li等[123]則嘗試在合成過程中添加2%(質量分數)LiF以降低H2O和CO2在LLZO中的擴散,以此來抑制Li2CO3的形成。LiF的添加使Li和石榴石之間的ASR降低至345 Ω·cm2。
在已報道的文獻中,Al2O3的原子層沉積(ALD)效果是最好的。該方法可以將室溫下的ASR從1710 Ω·cm2降低至1 Ω·cm2[112],效果十分顯著。在ALD處理過的LLZO表面上熔融金屬Li時,Al2O3層的存在增加了界面接觸面積,同時抑制了Li2CO3的形成。盡管ALD非常有效,但由于較高的成本和操作的復雜性,ALD法在商業應用中并不普及。簡單的拋光可能對于實際應用來說更具吸引力。Thangadurai等[122]采用濕法處理技術,將Zn(NO3)2溶液均勻分布在LLZO表面并使其熱分解形成ZnO,同樣有效地降低了Li和LLZO之間的ASR。碳的引入被認為可以有效改善Li與固態電解質的界面接觸性。Duan等[119]采用的是Li-C復合負極。Li-C復合材料是通過在250oC的熱板上向連續攪拌熔融的Li中添加石墨粉制備的。Li-C復合材料與石榴石的界面電阻僅為11 Ω·cm2,與Li-LLZO界面電阻381 Ω·cm2相比有了很大的降低。
正極材料與LLZO界面的構筑目前研究得較少。為確保LLZO和正極材料的界面有良好的接觸,研究人員進行了各種嘗試。Ohta等[124]通過脈沖激光沉積技術,在Li6.75La3Zr1.75Nb0.25O12上沉積了LiCoO2,在3.95 V(vs.Li/Li+)充電狀態下正極-電解質界面處的ASR為170 Ω·cm2,與液態有機電解質的ASR值相當。Liu等[125]將V2O5涂覆在Li7La2.75Ca0.25Zr1.75Nb0.25O12顆粒上,隨后將其加熱至800oC。該電解質的室溫ASR降低至71 Ω·cm2。Park等[126]也同樣采取了類似的退火技術,把LiCoO2在LLZO表面加熱至700oC,保溫6 h。飛行時間二次離子質譜(TOF-SIMS)和XRD研究證實,在加熱時,LiCoO2的存在會導致c-LLZO相轉變t-LLZO。除了傳統的插層正極材料外,研究人員同樣將目標放在與下一代正極材料的界面,特別是與硫的界面的構筑方面。Fu等[127]的實驗結果表明LLZO與熔融硫混合后仍為立方相,但計算和實驗結果表明,在硫與LLZO界面上形成了由Li2S和Li2SO4組成的鈍化層。
為了實現全固態鋰電池的長循環,在構筑電池單元時,研究人員通常會在固態電解質和正/負極之間添加“少量”(通常為幾μL)傳統液體電解質。研究發現,盡管具有“混合電解質”的電池顯示出比全固態電池更高的容量,但其循環能力比傳統液體電池更差。Xu等[128]認為混合電解質電池的不良循環可能是由于與液體電解質接觸的石榴石表面上的Li+/H+交換和固液電解質高電阻界面的形成造成的。Liu等[129]證實了Li6.5La3Zr1.5Ta0.5O12對于典型的鋰離子電池液態電解液LP30是不穩定的。由于二者間的反應,在其接觸界面上會生長一層新的SEI界面層,該SEI主要由Li2O、Li2CO3、LiF以及一些有機物等構成,大大增加了界面的電阻。缺少少量的傳統液體電解質的添加,全固態電池很難進行充放電循環。雖然從商業電池中去除液體電解質并過渡到全固態電池是理想的,但石榴石和正極材料之間的高界面電阻仍然是一個難以解決的問題。在未來的電池設計中,液體電解質可能仍會保留一段時間。綜合來看,安全性高、能量密度潛力大的固態電池必然是下一代動力電池的研發方向,但截止到目前,確實還沒有任何量產的產品能夠在各方面明顯勝過傳統鋰離子電池。固態鋰電池更大的能量密度空間成為一些企業追求固態電池的一個重要原因。就目前情況來看,單純的LLZO陶瓷片是無法用來制備大容量電芯的。除此之外,LLZO基固態鋰電池成本也遠高于液態鋰電池;在工程化層面,全固態電池在材料和工藝層面也面臨很多問題,比如充放電過程中的膨脹問題,加壓、涂布等生產工藝的挑戰,這些都是商業化生產中必須解決的問題。
雖然LLZO基固態電解質用于固態電池商業化之路困難重重,但是國內外眾多企業紛紛入局固態電池研發。2011年,法國博洛雷(Bolloré)首次將商業化固態電池應用于EV。2018年,本田、日產聯手豐田、松下、湯淺以及日本三大化工集團成立了“鋰電池技術與評估中心”,以聯盟的形式開展固態電池的研發。2018年11月,清陶新能源宣布建設第一條單體能量密度達到400 W·h/kg的固態電池生產線,并于2019年11月正式投產。一直堅持混動和氫燃料電池路線的豐田計劃在2020年以后全面引進EV,在2020年東京奧運會期間推出一款搭載固態電池的電動汽車,預計2025年左右可以大規模生產固態電池汽車。寶馬集團正與固態電池公司Solid Energy合作共同開發固態電池,大眾集團同樣看好固態電池前景,并入股研發固態電池的創業公司Quantum Scape。此外,從2019年5月起,日本政府將出資16億日元,聯合國內豐田、本田、日產、松下、GS湯淺、東麗、旭化成、三井化學、三菱化學等大型汽車廠商、電池和材料廠商,共同研發固態電池。
5 結 語
電解質作為電池的重要組成部件,其性能的好壞直接決定了電池性能的優劣。總體而言,與液態電解液相比,固體電解質在材料安全性、穩定性和組裝電池的設計簡單性等方面具有明顯優勢。但固態電解質體系仍然面臨離子電導率較低(對比電解液)以及固-固界面不兼容的問題。LLZO作為最具市場化潛力的固態電解質材料之一,一直吸引著眾多研究人員的關注,通過深入了解LLZO晶體結構以及通過元素摻雜對富鋰石榴石結構進行優化,已經將LLZO的鋰離子電導率提高一個數量級,并且諸多結果表明,對富鋰石榴石家族的晶體結構和Li+濃度的調控已經達到頂峰。盡管如此,構筑低阻抗與重現性高的固體電極/固體電解質界面等突出問題仍有較長的路要走,固態電池的春天還沒有到來。從綜合布局固態電池的企業數量以及電動汽車產業需求來看,固態動力電池產業仍然是風險與機遇并存,并且存在潛在風險難以評估的問題。
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
