固相萃取—微波衍生化—GC-MS法同時測定水中6種雌激素物質
2020-04-25 04:59:42袁宏宇王欣澤遲莉娜字建婷王榮會
化工環保 2020年2期
袁宏宇,王欣澤,,沈 劍,遲莉娜,字建婷,王榮會
(1. 上海交通大學 環境科學與工程學院,上海 201100;2. 上海交通大學 云南(大理)研究院,云南 大理 671000)
環境雌激素(Environmental estrogens,EEs)是屬于環境內分泌干擾物的一類化學物質,進入生物體會破壞機體穩定性和正常調控作用[1],包括天然內源雌激素和人工合成外源雌激素兩大類。天然內源雌激素通過動物及人體排泄物釋放到環境中,包括雌酮(E1)、17β-雌二醇(β-E2)、17α-雌二醇(α-E2)、雌三醇(E3)[2]等。化學工業技術的飛速發展導致大量合成有機化學品釋放到環境中,其中17α-乙炔基雌二醇(EE2)、雙酚A(BPA)是具有內分泌干擾效應的典型雌激素類物質[3-5]。大量研究表明,雌激素類物質在濃度極低(ng/L級別)情況下就能對生物體的內分泌系統等造成損傷,且雙酚A、EE2與天然雌激素之間可能還會存在協同效應[6-7]。
氣相色譜-質譜聯用方法(GC-MS)因其分離效果好、靈敏度高及成本相對較低等優點成為目前最為常用的EEs分析測定方法[8-10],由于EEs具有痕量特征,通常需要經過過濾、富集及濃縮等復雜前處理步驟,從而給分析測定帶來了極大困難。目前常用的前處理方法包括固相萃取(SPE)[11-12]、液液萃取(LLE)[13-14]、固相微萃取(SPME)[15]等,其中固相萃取因其簡便高效等優點得到廣泛應用。
本工作系統優化了氣相色譜柱升溫程序、衍生化條件以及固相萃取小柱類型,建立了快速同時測定水中這6種雌激素物質的分析方法,對研究水體環境雌激素內分泌干擾效應具有重要應用價值。
1 實驗部分
1.1 儀器和設備
TRACE1300型GC,ISQ Series Quadrupole 型MS氣相色譜質譜聯用儀:美國ThermoFisher公司;TriPlus RSH型自動進樣器:美國ThermoFisher公司;SQP型電子天平:德國賽多利斯公司;SBAB-57044型CNW 12位固相萃取裝置:上海安譜實驗科技股份有限公司;HC-C18型、Poly-Sery HLB型、Poly-Sery PSD型、ENVI-18 SPE型等4種萃取小柱:上海安譜實驗科技股份有限公司。
1.2 試劑及標準溶液配制
雌激素分析標準物(E1、17α-E2、17β-E2、E3、EE2、BPA)、標準替代物(17β-E2-2,4-d2)以及內標物(滅蟻靈):均為分析標準品,均購自阿拉丁試劑有限公司;含有1% (w,下同)三甲基氯硅烷(TMCS)的雙(三甲基硅烷基)三氟乙酰胺(BSTFA):購自上海麥克林公司。
甲醇:色譜級,純度≥99.9%;乙酸乙酯:HPLC級,純度≥98.0%;正己烷:HPLC級,純度≥98.0%;無水吡啶溶液:純度≥99.8%。
精確稱量6種目標物質的標準品以及標準替代物各50 mg,分別溶于50 mL甲醇中,配制成1 g/L的高濃度單標溶液,分別移取100 μL到10 mL容量瓶中,再加入甲醇配制成10 mg/L的混合標準溶液。準確稱量50 mg 滅蟻靈溶于50 mL 正己烷中,配制成1 g/L 的高濃度內標溶液。配制好的溶液放置于-20 ℃冰箱中冷凍避光保存,根據需要逐級稀釋使用。
1.3 實驗方法
1.3.1 固相萃取
取200 μL各雌激素質量濃度分別為1 mg/L的混合標準溶液加入到1 L超純水中,配成雌激素質量濃度分別為200 ng/L的水樣。選擇HC-C18、Poly-Sery HLB、Poly-Sery PSD以及ENVI-18等4種不同類型的萃取小柱對水樣進行固相萃取,考察不同萃取小柱對6種雌激素物質的回收率。每組實驗重復3次,計算平均回收率及標準偏差。
固相萃取分為以下4個步驟:1)萃取小柱活化與平衡,先分別取5 mL乙酸乙酯與5 mL甲醇過柱,使萃取小柱活化,再用5 mL超純水清洗2次,流量約為2 mL/min;2)水樣過柱,用真空泵抽取過濾水樣通過萃取小柱,富集待測物于填料中;3)淋洗雜質,用10 mL 10%(w)甲醇溶液洗滌小柱,抽真空30 min左右直至小柱表面干裂;4)洗脫目標物,用10 mL乙酸乙酯洗脫萃取小柱中的待測物,控制流量為2 mL/min左右,并用試管收集洗脫液。
1.3.2 衍生化
將固相萃取洗脫液(衍生化優化實驗時以配制的雌激素混合標準溶液代替固相萃取洗脫液)在30 ℃條件下通入溫和高純氮氣,干燥完全后加入25 μL(BSTFA+1% TMCS)+50 μL吡啶作為衍生化試劑,一定微波功率下加熱一定時間,再在30 ℃條件下用溫和氮氣干燥,加濃度為1 mg/L的內標物滅蟻靈溶液定容至400 μL,轉入2 mL進樣瓶,待GC-MS分析。
1.3.3 GC-MS
1)GC條件:TR5-MS型石英毛細管色譜柱(30 m×0.25 mm×0.25 μm);99.999%高純氦氣為載氣,采用恒流模式(載氣流量1.2 mL/min);不分流進樣,每次取樣量為1.0 μL;隔墊吹掃5.0 mL/min。實驗考察色譜柱升溫程序。
2)MS條件:電子轟擊離子源(EI),電離電壓70 eV;離子源溫度250 ℃,傳輸線溫度280 ℃;溶劑延遲時間10 min。采用全掃描模式進行定性分析、離子掃描模式進行定量測定,全掃描質量數范圍(m/z)為33~650。使用Xcalibur軟件進行定性分析,確定6種雌激素目標物、標準替代物及內標物所對應的標準譜圖以及保留時間,選擇3~4個相對豐度較強、分子量較大的碎片離子作為定性離子,并選擇其中豐度最強且與其他物質特征離子沒有干擾的定性離子作為定量離子。
1.3.4 標準曲線、方法檢出限及線性范圍的確定
用含有6種目標物的混合標準溶液配制成各物質含量分別為1,3,5,10,25,50,100,200,300 ng的梯度濃度系列標準溶液,再分別加入物質含量為100 ng的17β-E2-2,4-d2(標準替代物)溶液,在優化條件下進行固相萃取—微波衍生化—GC-MS分析測定,以各目標物的定量離子峰與內標物的定量離子峰的峰面積比值為縱坐標Y,以各目標物質量濃度X(μg/L)為橫坐標進行線性回歸分析。以信噪比S/N =3結合目標物回收率計算分析方法的檢出限(LOD),以信噪比S/N=10計算該方法的定量限(LOQ)。
2 結果與討論
2.1 色譜柱升溫程序的選擇
氣相柱溫箱的升溫程序影響著目標物的保留時間、峰分離度以及峰形等,對定性與定量分析均會產生一定的影響。設置3種不同的升溫程序(見表1),考察全掃描色譜圖出峰效果。實驗結果表明,程序升溫方式2條件下雌激素物質的全掃描總離子色譜譜圖中6種標準物質及內標物均出峰、峰形良好且分離度較好,見圖1。因此確定柱溫箱最佳升溫程序為:初始溫度50 ℃,保持2 min;以20 ℃/min的升溫速率上升至260 ℃,保持5 min;再以3 ℃/min的升溫速率上升至280 ℃,保持5 min。在此升溫程序條件下,總掃描時長為24.62 min。

表1 3種程序升溫方式
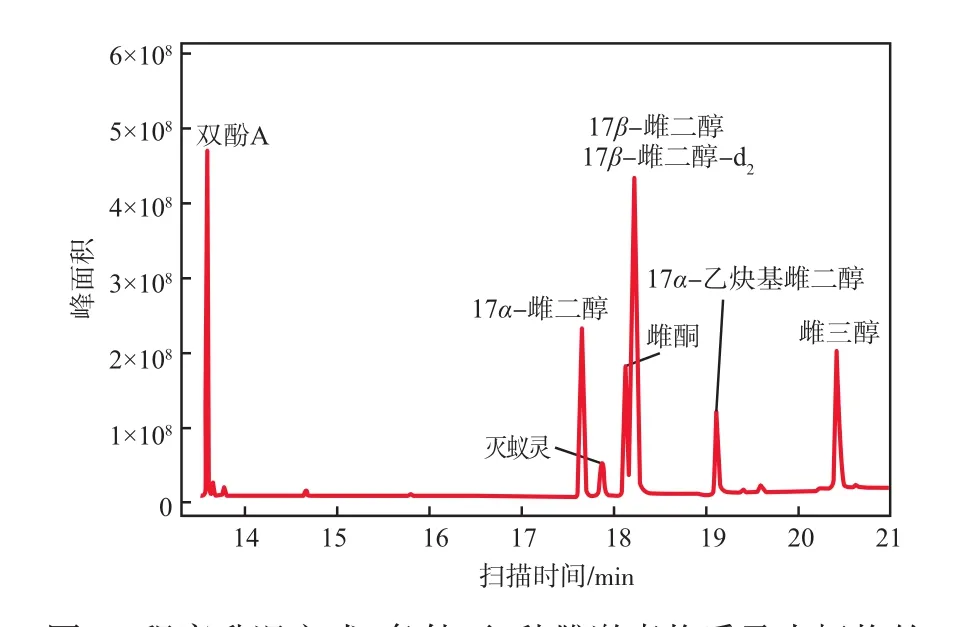
圖1 程序升溫方式2條件下6種雌激素物質及內標物的全掃描總離子色譜譜圖
2.2 色譜與質譜特征
根據圖1數據進行定性分析,得到每種雌激素物質和內標物的保留時間及特征離子,見表2。
2.3 衍生化條件優化
由于雌激素類物質為弱極性至中等極性化學物,毛細管色譜柱為非極性柱,目標物質直接進樣的出峰效果差,且會對色譜柱造成損害,故采用BSTFA+1%TMCS+吡啶作為衍生化試劑。目標物經衍生化后,產物單一穩定,分離效果好,靈敏度高[16-17]。
2.3.1 衍生化加熱方式的選擇
因可通過提高極性分子運動速度和碰撞頻率快速加熱物質、快速提高物質之間的化學反應速率,微波在樣品前處理中得到了廣泛應用[18-21]。為了提高衍生化效率,本實驗將微波加熱衍生化與水浴及烘箱加熱衍生化的效果進行了對比研究。預實驗結果表明:水浴及烘箱加熱衍生化的最佳條件為40 ℃反應20 min,這與王園園等[22]的研究結果一致,加熱溫度過高可能會生成不利于GC-MS分析的副產物,延長反應時間,對提高目標物回收率貢獻不大。衍生化方式對雌激素物質峰面積的影響見圖2。由圖2可見:常溫20 ℃條件下峰面積最小,說明衍生化效果最差;水浴40 ℃和烘箱40 ℃加熱條件下峰面積大致相當;微波252W加熱3 min衍生化的各目標物峰面積略低于水浴和烘箱加熱;微波567 W加熱3 min的衍生化效果最佳,各目標物的峰面積相比常溫20℃衍生化增加了24.09%~42.97%,相比烘箱加熱衍生化增加了12.12%~24.31%,相比水浴加熱衍生化增加了8.06%~33.53%,相比微波252 W加熱3 min衍生化增加了16.62%~28.12%。微波加熱使衍生化時間大幅縮短至3 min,與傳統衍生化方式相比,衍生化效率顯著提升。
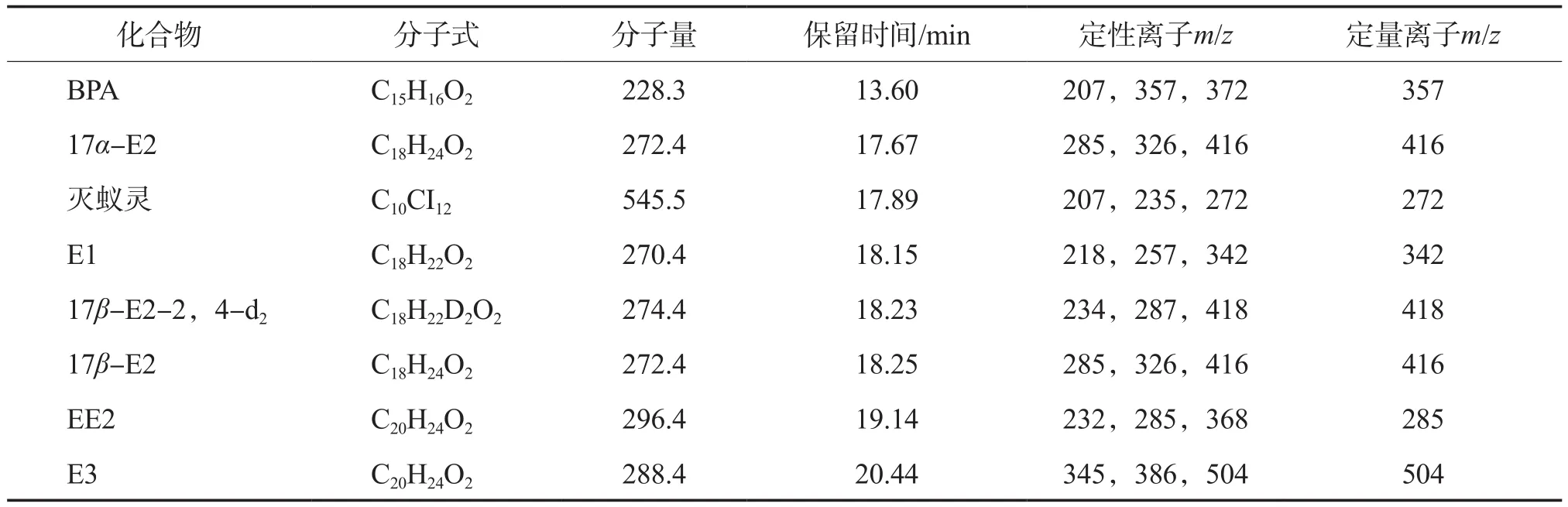
表2 雌激素物質和內標物的保留時間及其特征離子
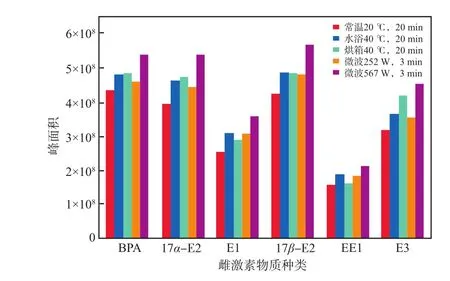
圖2 衍生化方式雌激素物質的峰面積
2.3.2 微波衍生化條件
微波功率和加熱時間是影響微波衍生化效果的關鍵因素[23-24]。采用BSTFA +1%TMCS+吡啶作為衍生化試劑,分別考察不同微波功率和不同加熱時間對6種雌激素物質衍生化效果的影響,每組實驗條件重復3 次,計算平均峰面積。
2.3.2.1 微波功率
在微波加熱時間為3 min的條件下,微波功率對雌激素物質峰面積的影響見圖3。由圖3可見:252 W微波加熱功率下,雌激素物質峰面積均較小,說明衍生化反應不完全導致衍生化效果差;微波功率升高至315 W時,雌激素物質峰面積均達到最大,說明衍生化效率達到最佳;微波功率調至406 W時雌激素物質峰面積明顯減小,再繼續提高微波功率也無明顯變化,說明微波功率過高時部分衍生化產物可能發生分解或與衍生化試劑進一步反應生成了不利于GC-MS分析的副產物,導致雌激素物質峰面積減小,衍生化效果變差。
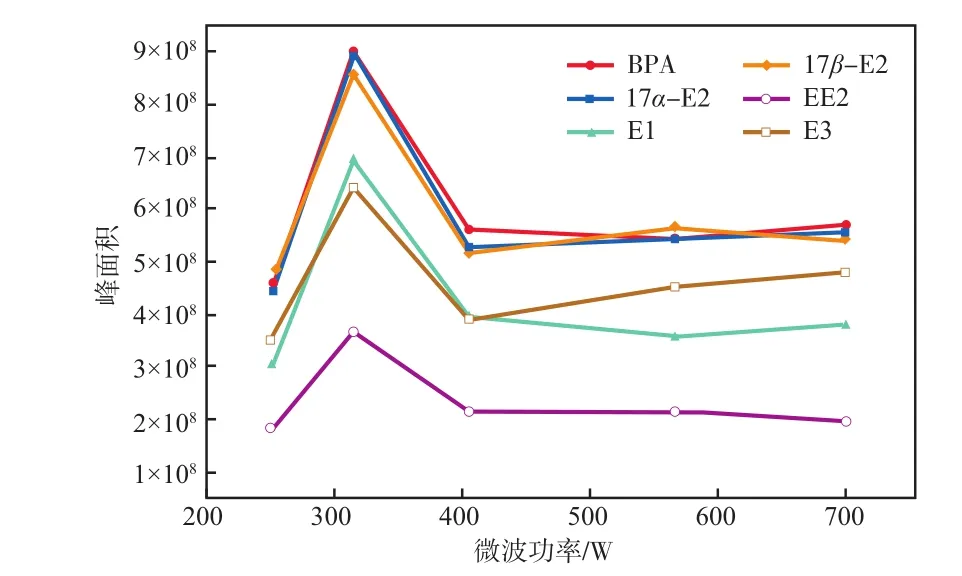
圖3 微波功率對雌激素物質峰面積的影響
2.3.2.2 微波加熱時間
在微波功率315 W條件下,微波加熱時間對雌激素物質峰面積的影響見圖4。
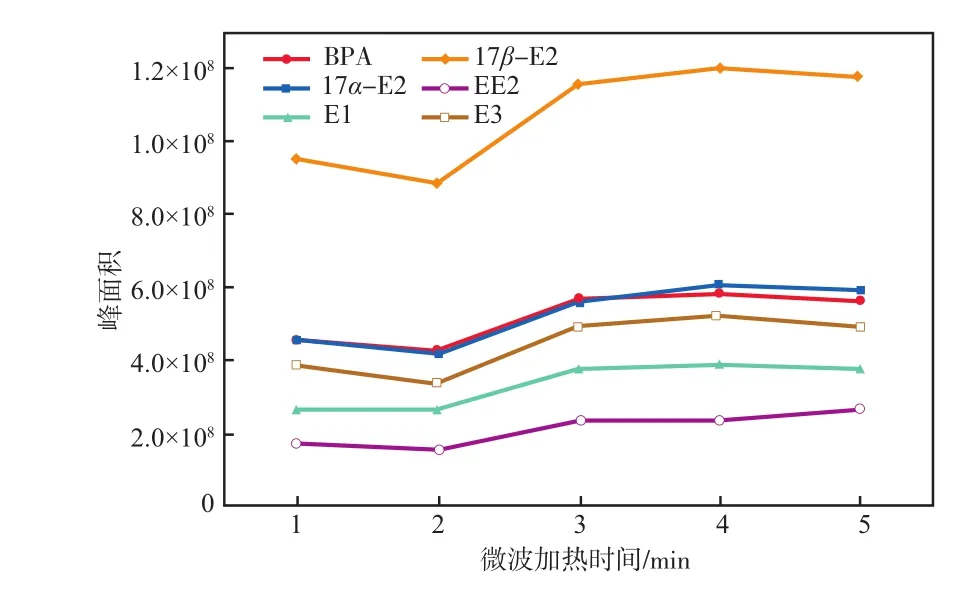
圖4 微波加熱時間對雌激素物質峰面積的影響
由圖4可見:隨著微波加熱時間增加,雌激素物質峰面積逐漸增大;當微波加熱時間為4 min時,6種雌激素物質的峰面積均最大,衍生化效果最好;再增加微波加熱時間則衍生化效果無明顯變化。為了防止衍生化產物分解并保證最佳衍生化效率,所以選擇微波功率315 W條件下加熱4 min為最佳衍生化條件。
2.4 固相萃取條件優化
4種不同類型萃取小柱吸附6種雌激素物質的回收率見圖5。
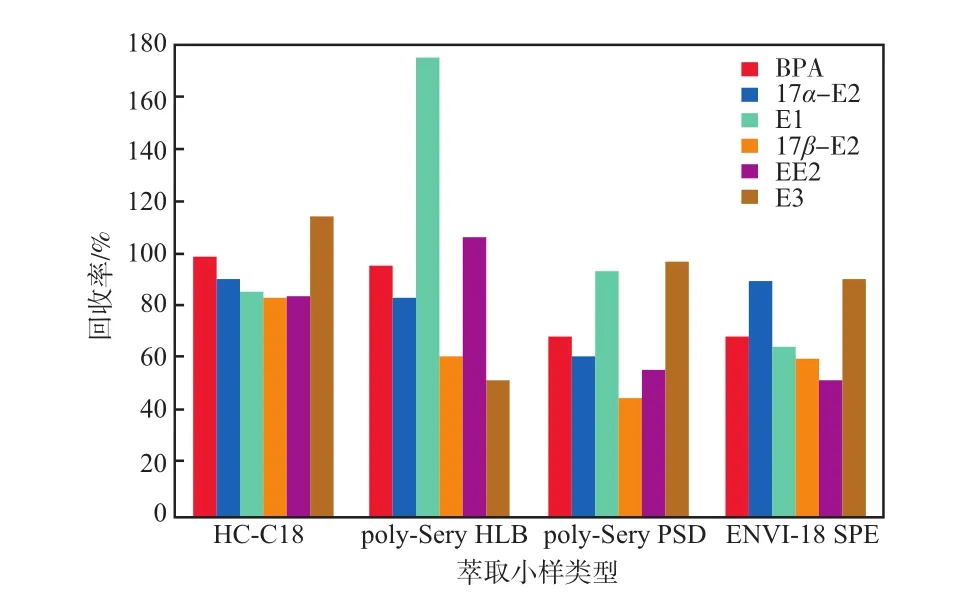
圖5 4種不同類型萃取小柱吸附6種雌激素物質的回收率
由圖5可見:HC-C18小柱對6種雌激素物質的回收率范圍為82.93%~115.00%,對6種雌激物質吸附效果良好;Poly-Sery HLB小柱對6種雌激素物質的回收率范圍為50.95%~174.14%,回收率波動范圍偏大,其中對E3和17β-E2吸附效果較差,回收率分別只有50.95%和60.51%;Poly-Sery PSD小柱對6種雌激素物質的回收率范圍為45.15%~93.10%,對E1和E3的回收率均高于90%,但對其余4種目標物的回收效果相對較差,均低于70%;ENVI-18 SPE小柱對6種雌激素物質的回收率范圍為52.21%~90.18%,對E3和17α-E2吸附效果較好,其余目標物則效果一般。綜上所述,HC-18小柱對6種雌激素物質的吸附效果比其他3種小柱好,且吸附穩定。
2.5 標準曲線、方法檢出限及線性范圍
6種雌激素物質的線性回歸方程、相關系數、檢出限、定量限及線性范圍見表3。由表3可見,各目標物峰面積與其質量濃度的線性相關性良好,相關系數(R)均大于0.999;BPA、E1、EE2及E3的線性范圍為3~300 ng/L,17α-E2及17β-E2的線性范圍為5~300 ng/L;方法檢出限為2.0~5.0 ng/L。
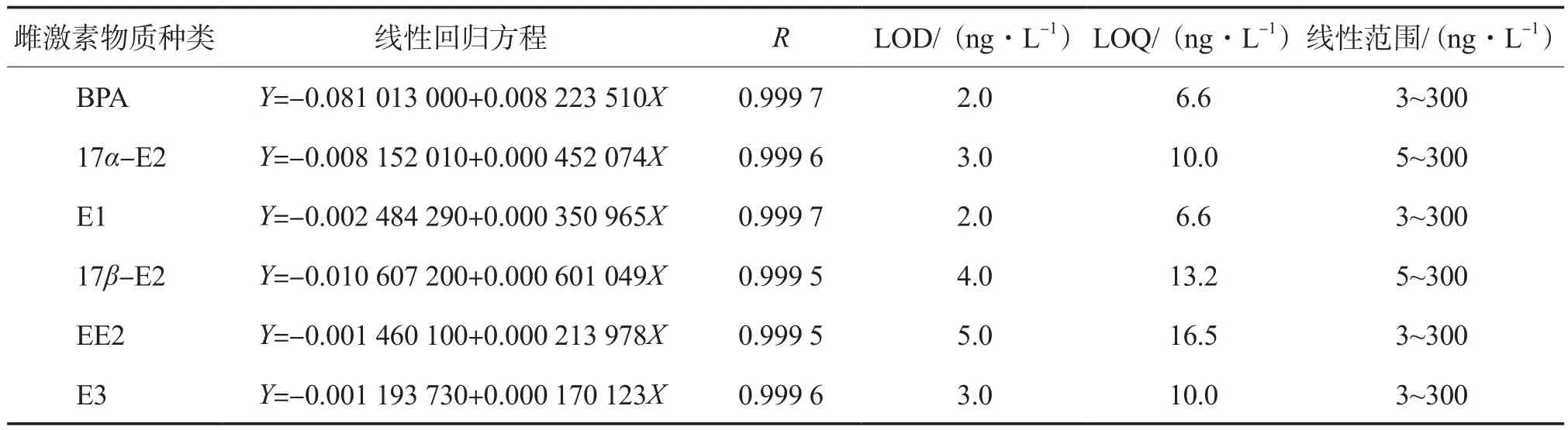
表3 6種雌激素物質的標準曲線及相關系數、檢出限、定量限及線性范圍
2.6 方法加標回收率及精密度
6種雌激素物質的回收率和相對標準偏差見表4。由表4可見,3種加標水平下的加標回收率范圍為76.45%~96.24%,相對標準偏差RSD范圍為3.2%~9.8%(n=3)。表明本方法的回收率較高,精密度良好。

表4 6種雌激素物質的回收率和相對標準偏差(n=3)
2.7 與其他方法的比較
本方法與其他方法的對比見表5。由表5可見:本方法可以同時測定6種雌激素物質,分析物數量比其他3種方法更多;采用微波加熱將衍生化時長縮短至4 min,相比其他方法,衍生化效率顯著提升;同時還能保證方法檢出限滿足水樣測定要求。綜上,本方法靈敏快捷、簡便高效,適用于大批量水樣雌激素的分析測定。

表5 本方法與其他方法的對比
2.8 實際水樣分析
采用建立的分析方法測定了云南大理洱海流域3處地表水樣,測定結果見表6。由表6可見:除17β-E2外,其余5種雌激素物質均被檢出,濃度范圍為7.24~128.64 ng/L;其中17α-E2的流域濃度最低,為7.24~10.88 ng/L;3處水樣中,水樣#285的E1濃度顯著高于其他兩處水樣,達到128.64 ng/L。
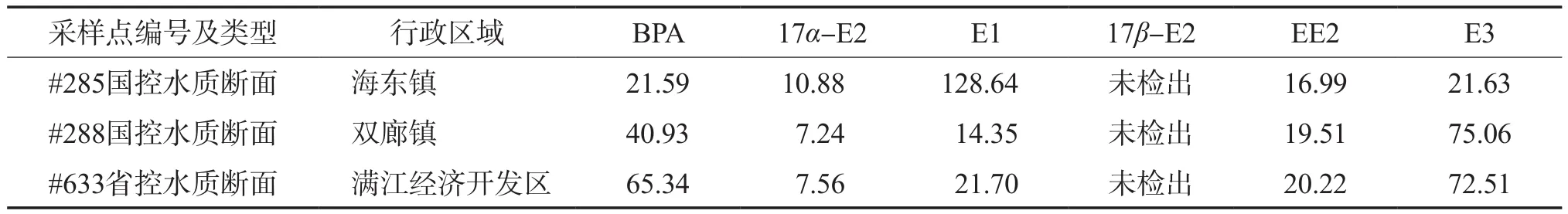
表6 實際水樣的分析結果 ρ,ng/L
3 結論
a) 通過對氣相色譜柱升溫程序、前處理步驟中的固相萃取及衍生化條件進行優化,建立了可快速同時測定天然雌激素物質雌酮、17α-雌二醇、17β-雌二醇、雌三醇以及人工合成雌激素17α-乙炔基雌二醇、雙酚A等6種雌激素物質的固相萃取—微波衍生化—GC-MS分析方法。
b)色譜柱最佳升溫程序為:初始溫度50 ℃,保持2 min;以20 ℃/min速率升至260 ℃,保持5 min;最后以10 ℃/min速率升至280 ℃,保持5 min。6種雌激素物質的全掃描色譜圖的出峰效果良好,且分離度高。
c)最佳衍生化條件為:在試樣中添加BSTFA+1%TMCS+吡啶作為衍生化試劑,315 W微波加熱4 min。相比烘箱、水浴等傳統衍生化加熱方式,微波加熱時間縮短至4 min,衍生化效率提升明顯。
d)最佳固相萃取小柱類型為HC-C18小柱,回收率范圍為82.93%~115.00%。本方法對BPA、E1、EE2及E3的線性范圍為3~300 ng/L;17α-E2及17β-E2的線性范圍為5~300 ng/L;方法檢出限為2.0~5.0 ng/L,加標回收率為76.45%~96.24%。
猜你喜歡
中老年保健(2021年12期)2021-11-30 02:58:01
兒童故事畫報(2019年5期)2019-05-26 14:26:14
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
中華詩詞(2018年11期)2018-03-26 06:41:34
Coco薇(2016年8期)2016-10-09 02:11:50
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國醫藥科學(2015年19期)2015-02-27 12:33:11
