三維網絡結構氧化銅納米線鋰離子電池負極材料的制備和性能研究*
2020-04-28 10:24:22曹宇光周佳盈ManonAssuncaoLourencoKevinPeterHomewood鮑鈺文夏曉紅
功能材料 2020年4期
曹宇光,肖 煌,周佳盈,高 云,Manon D’Assuncao Lourenco,Kevin Peter Homewood,鮑鈺文,夏曉紅
(湖北大學 材料科學與工程學院,功能材料綠色制備與應用教育部重點實驗室,有機化工新材料湖北省協同創新中心,武漢 430062)
0 引 言
隨著便攜式電子設備、儲能設備和電動汽車的飛速發展,對更小,更輕,容量更大的可移動電池的需求迅速增加[1-3],基于石墨材料的傳統鋰離子電池(LIB)已不能滿足未來耗能設備不斷增長的需求,低容量、倍率能力不足的電極材料嚴重限制了現有電池的能量和功率密度[4-6]。開發具有更高能量密度、更好倍率性能以及更長使用壽命的新型電極材料成為鋰離子電池未來發展的重要方向[7]。
過渡金屬氧化物具有比石墨(372 mAh/g)更高的比容量,是下一代LIB最有潛力的負極材料之一[8-9]。CuO作為1種p型半導體,具有帶隙窄(1.2 eV)[10]、理論比容量高(670 mAh/g)、成本低廉、易于合成及環境友好等優勢[11-13],被認為是一種非常具有應用前景的LIB負極材料。CuO用作鋰離子電池負極材料的工作機理與其他金屬氧化物一樣,可以概括為:MO+2Li++2e-←→Li2O+M(M=Cu,Fe,Ni,Co等)。在上述電化學轉化反應過程中,會因鋰嵌入/脫出出現巨大的不均勻體積變化(約174%),導致活性材料的結構破裂和容量衰減[14-15],從而表現出較差的循環穩定性[16-18]。制備CuO微/納米結構是解決體積變化問題的有效途徑之一[19-20],如C.Wang等通過溶液法合成了CuO納米微球[21];R.Zhang等通過電沉積法在銅泡沫上合成了CuO納米線陣列[22];W.Yang等通過電化學氧化在銅泡沫上包覆三維氧化銅納米片[23];這些納米結構的CuO保持了較高的活性,提供了有效的間隙結構來減輕鋰離子嵌入/脫出過程中的體積變化,可以有效改善CuO負極材料的體積變化問題[24-25]。目前CuO納米結構應用于鋰離子電池的性能總結如表1所示,電池的循環壽命和比容量還有待進一步提升,開發具有高穩定性,高循環壽命的CuO基鋰離子電池具有重要的實際應用價值。
表1 各種CuO鋰離子電池負極材料的性能比較。
Table 1 Summary of LIB made from CuO anode materials
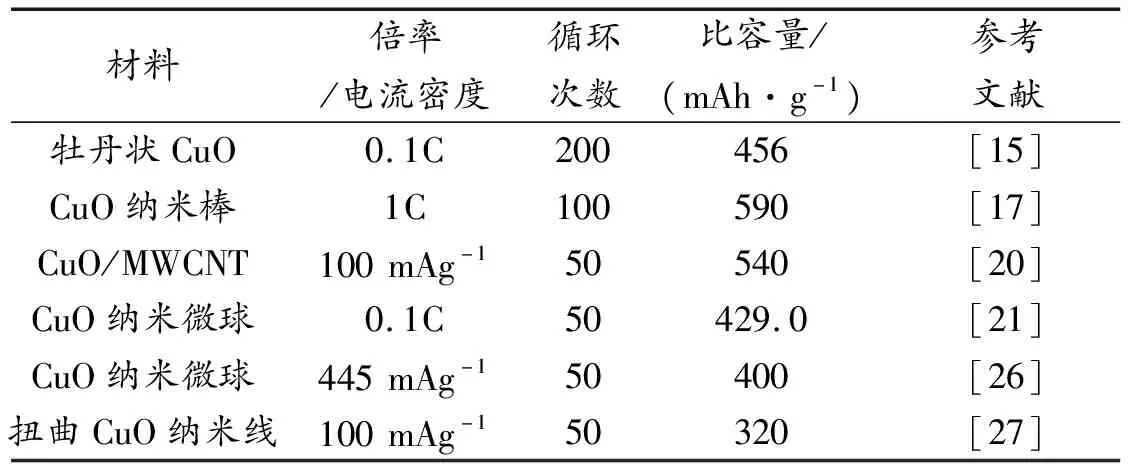
材料倍率/電流密度循環次數比容量/(mAh·g-1)參考文獻牡丹狀CuO0.1C200456[15]CuO納米棒1C100590[17]CuO/MWCNT100mAg-150540[20]CuO納米微球0.1C50429.0[21]CuO納米微球445mAg-150400[26]扭曲CuO納米線100mAg-150320[27]
本文采用陽極氧化法及退火處理在銅箔上制備出三維交聯結構氧化銅納米線,將其直接用作鋰離子電池負極材料,無須粘結劑和其他任何添加劑,簡化了LIB的組裝方法。相比于垂直生長納米線陣列,交錯的三維網絡結構納米CuO表現出非常強的穩定性,500圈循環后仍保持高的可逆容量和良好的倍率性能。
1 實驗部分
將9 μm厚的銅箔(99.9%)依次用稀鹽酸、丙酮和乙醇進行預處理,將處理過的銅箔作為工作電極,在電化學工作站進行陽極氧化反應,Ag/AgCl電極為參比電極,鉑電極為對電極,電解液為1 mol/L的氫氧化鉀溶液,固定氧化電壓為0.6 V,氧化時間分別為600、800、1000和1 200 s。
反應完成后將銅箔取出,用去離子水清洗3次,自然干燥后將其置于管式退火爐中,在空氣氣氛下,以3 ℃/min的升溫速率升溫至200 ℃,保持4h后自然降溫至室溫,得到黑色負極材料。使用Sigma500場發射掃描電子顯微鏡(卡爾·蔡司)對負極材料進行形貌分析,D8 Advance X射線衍射儀(德國布魯克)對樣品進行結構表征,LabRAM HR Evolution拉曼光譜儀(HORIBA)對樣品進行拉曼表征。
將退火后的樣品沖成電極片后放于Lab2000手套箱內(伊特克斯),鋰片作對電極與之匹配,使用CR2016型紐扣電池殼(科路德)進行組裝,電解液為SW2001A型號的商用LiPF6電解液(EC比DEC體積比為1:1)(珠海賽緯電子材料)。采用新威電池測試系統(BTS-5V 10mA)(新威電子)對半電池進行充放電性能測試,使用CHI660E電化學工作站(辰華)對半電池進行電化學性能測試。
2 結果與討論
圖1(a)為沉積時間為1 000 s的樣品的XRD圖譜。對比CuO標準PDF衍射峰(JCPDS: 44- 0706)可以看出,在2θ=35.2°和38.5°出現的峰分別對應CuO的(-111)、(111)晶面。衍射峰發生寬化,說明樣品尺寸較小,根據(111)衍射峰所對應的半高寬采用謝樂公式計算晶粒尺寸為35.2 nm。在2θ=43.3°、50.4°所出現的強峰來自于銅箔基底的(111)和(200)晶面。XRD結果表明采用陽極氧化和后退火處理可以成功制備結晶性能良好的CuO。圖1(b)為該樣品的拉曼光譜圖。CuO是單斜結構,總共有12種光-聲子振動模式,4Au+5Bu+Ag+2Bg,其中3種Ag+2Bg具有拉曼活性[28]。圖1(b)中顯示的在286、335、618和1141cm-1處的拉曼峰分別對應氧化銅的Ag,Bg,Bg和2Bg的震動模式[29],也證明了退火后的樣品為CuO。且圖中所示的拉曼峰都具有較大的半高寬,說明CuO的尺寸較小,與XRD衍射峰的寬化一致。
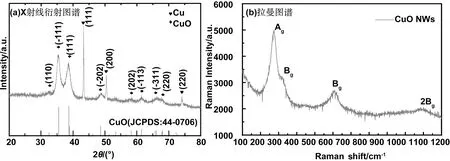
圖1 陽極氧化時間為1 000 s的樣品的(a)X射線衍射圖譜;(b)拉曼圖譜
圖2為不同氧化時長制備的氧化銅樣品的場發射掃描電子顯微鏡照片。從圖2(a)可以看出,較短氧化時間(600 s)制備的樣品為傾斜于基底,交錯生長,均勻覆蓋于銅箔表面的束狀納米棒。隨著反應時間的增加,納米棒逐漸變粗變長。從圖2(b)和(c)可以看出,800 s以上的氧化時間所制備的樣品由棒狀轉變為粗細較為均勻的線狀,且密度逐漸增加,線的頂端逐漸彎曲,在不同的納米線束之間搭建成三維網絡結構。氧化時間為1 200 s制備的樣品尺寸進一步增大,逐漸形成簇狀,但是所制備的納米線有脫落現象,反應后溶液呈現淡藍色,過長的納米線導致其與基底間的接觸變弱。
將所制備的CuO納米線直接組裝成半電池進行電化學測試,由于CuO直接在銅箔上生長,銅箔可以用來作為鋰離子電池的集流體,因此CuO納米線作為負極材料與銅箔集流體之間無需添加粘結劑,大大簡化了電池的制作工藝,降低了電池的生產成本。圖3為不同反應時長制備的樣品組裝成電池后,在1C倍率下進行50圈恒流充放電測試的循環容量圖。陽極氧化時間分別為600、800、1 000、1 200 s的樣品作為負極材料的半電池首圈放電比容量分別為708、851、1172、969 mAh/g,初始的可逆比容量分別為479、527、594、558 mAh/g。循環50圈以后,4個樣品的可逆容量保留率分別為94.9%、100%、106.4%和111.6%。從圖中可以看出4個樣品50圈恒流充放電性能非常穩定,幾乎沒有衰減,相反800、1 000、1 200 s制備的樣品比容量在測試過程中還逐漸增加。1 000 s和1 200 s制備的樣品循環性能較為相似,充放電比容量明顯高于600 s和800 s,且1 000 s的樣品性能略高于1 200 s的樣品。

圖1 不同陽極氧化時長制備的氧化銅樣品的掃描電子顯微鏡照片
600 s和800 s的樣品,由于生長時間較短,納米線的密度較低,鋰離子嵌入/脫出的量相對較少,所以二者的可逆比容量相比于1 000 s和1 200 s的樣品較低。隨著氧化時間增加,納米線繼續生長,體積逐漸增大,1 000 s和1 200 s的樣品具有更大的比表面積,鋰離子嵌入/脫出的量更多,表現出了較大的可逆比容量。

圖3 陽極氧化時間分別為600s,800s,1000s和1200s制備的氧化銅納米線作為負極材料的鋰離子電池在1C倍率下50次循環壽命測試曲線
從圖3還可以看出,4個樣品在實驗中均出現了比容量在初始下降后又逐漸增加的現象,這可能歸因于:(1)氧化物與基體之間的界面相互作用,(2)在連續充電步驟中不可逆的Li2O逐漸分解為氧化物和Li+。CuO的導電性很差,會嚴重限制Li+在活性物質表面的傳輸和反應[30]。而在放電過程中會形成納米尺寸的Cu顆粒,其尺寸會隨著循環次數的增加而逐漸減小,這些Cu顆粒均勻地分散在CuO納米線中,可以顯著改善其導電性,促進Li+在活性物質中嵌入和脫出[11,31],因而電池的比容量逐漸增加。
圖4為1 000 s和1 200 s的樣品作為負極材料組裝成電池后,在1C倍率下進行500圈恒流充放電測試曲線。從圖中可以得知,300圈以前,二者的可逆比容量相似,都沒有明顯的衰減,300圈以后,1 000 s的樣品持續維持在600 mAhg-1以上,500圈時的可逆比容量為607.6 mAhg-1,可逆容量保留率為102.3%;1200s的樣品在300圈以后比容量開始逐漸衰減,在循環500圈時,其可逆比容量的保留率為72.2%。1000s的樣品具有最好的可逆比容量以及循環性能。
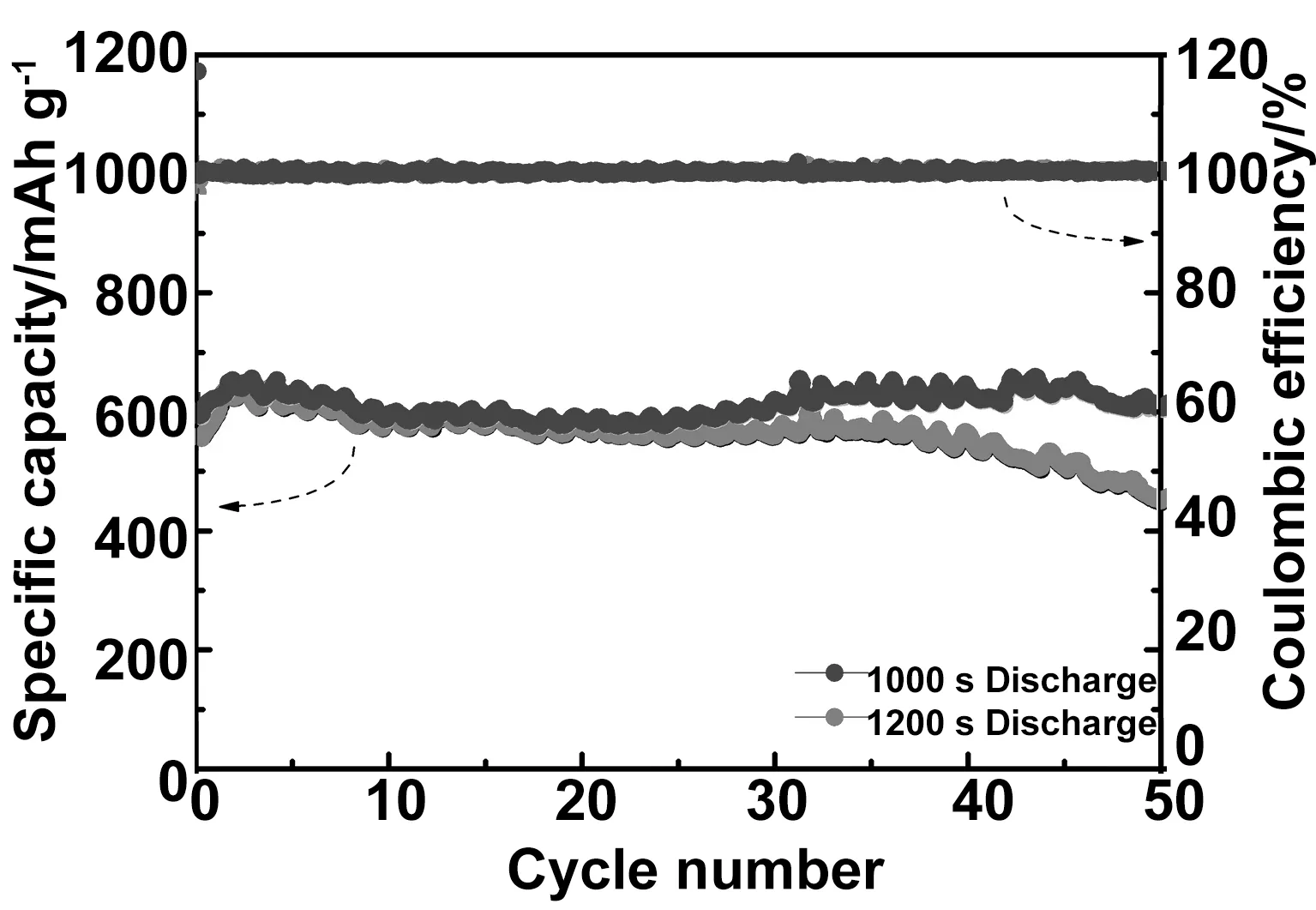
圖4 陽極氧化反應1000s和1200s的氧化銅納米線在1C倍率下的500次循環壽命測試曲線
對1 000 s制備的樣品電池進行電化學測試,圖5(a)為其在0~3 V電位范圍內以0.1 mVs-1速率掃描得到的CV曲線圖。由于首圈充放電過程中SEI膜形成,因此第一圈循環和隨后循環中的CV曲線不能完全重合。在隨后的CV曲線中,于2.29、1.39、0.65 V處出現3個陰極峰。在2.29 V處出現的第一個寬峰對應于伴隨CuO形成CuOⅡ1-xCuIxO1-x/2(0≤x≤0.4)固溶體的過程,在1.39 V處的第二個峰對應于Cu2O的形成,最后一個0.65 V處的陰極峰對應于Cu2O還原的Cu和Li2O的混合物[20]。同時,這些陰極峰伴隨著SEI膜的形成[32]。相對地,在1.53 V和2.54 V處出現的陽極峰分別表示的是Cu氧化為Cu2O以及Cu2O氧化為CuO的過程,同時也包括SEI膜分解的過程。
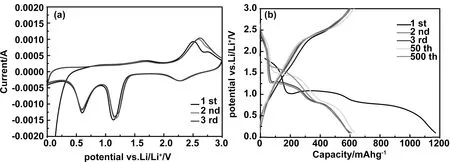
圖5 (a)1 000 s樣品半電池在0~3 V vs Li/Li+之間以0.1 mV/s的速率掃描的典型CV曲線;(b)1 000 s樣品陽極以1C倍率在0~3 V的電位范圍內的第1,第2,第3,第50和第500圈循環的充放電曲線
圖5(b)是1 000 s制備的CuO樣品以1C倍率在0~3 V之間的第1、第2、第3、第50和第500圈循環的充放電曲線。第一圈的放電和充電比容量分別為1172和595.3 mAh/g,對應的庫倫效率為50.8%,其中49.1 %的不可逆容量損失可能歸因于SEI膜的形成和電解質的分解[18,23]。同時,從圖中可以看出3個放電平臺分別位于2.2~2.5、1.2~1.4和0.6~0.8 V 3段電位范圍之間,對應于CV曲線中的3個陰極峰。此外,除去第一個放電曲線外,隨后的第2和第3圈循環放電和充電曲線彼此重合,在第50圈和第500圈循環時,放電電位有所遷移,充電曲線相互重合,整體表現出高度可逆的電化學性能。
圖6為1 000 s樣品的半電池在不同倍率下進行恒流充放電測試的比容量。在0.1、0.2、0.5、1、3和5 C倍率下對應的平均充電比容量分別為663.4、665.6、601.6、545.6、463.1和427.7 mAh/g,當倍率重新降低到0.1 C時,其平均充電比容量可達到643.9 mAh/g,接近在0.1C倍率下初始10圈循環的平均充電比容量,證明了樣品具有良好的可逆性能。值得注意的是,在5C的倍率下經過60圈循環,電池的充電比容量仍然可以維持在400 mAh/g以上。相比于其它CuO負極材料(表1),可以看出,這種交聯結構的CuO納米線具有更高的可逆比容量、更好的倍率性能和循環壽命。
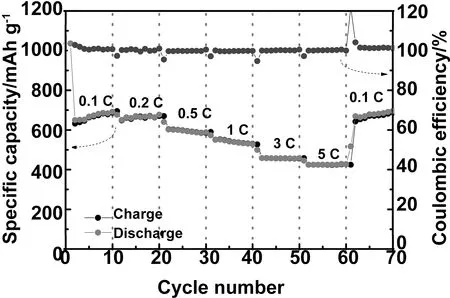
圖6 不同倍率下的1000s-CuO樣品半電池的比容量:0.1 C, 0.2 C, 0.5 C, 1 C, 3 C和5 C
圖7為1 000 s和1 200 s制備的樣品在1 C倍率下循環500圈后的SEM形貌照片。和圖2(c)對比發現,1 000 s制備的CuO納米線尺寸明顯增大,說明在充放電過程中體積變化仍然存在。500圈循環后納米線間三維交聯結構更加明顯,納米線間具有大的孔隙率,納米線之間相互支撐保持穩定的機械性能,從而可以保證電池具有良好的循環壽命。從插入的放大圖可以看出,CuO納米線的表面出現多孔結構,但是整體仍然保持完整的線狀結構。而1200s制備的樣品由圖2(d)中的發射狀納米棒簇坍塌為圖7(d)中的團聚塊狀結構,納米棒間間隙大幅減少,雖然插入圖中CuO保持單根棒狀結構,但是整體結構已坍塌,團聚的塊狀結構減少了鋰離子與活性物質的接觸面積,使鋰離子的嵌入/脫出量減少,因此電池300圈后的循環性能逐漸衰減。

圖7 (a)1 000 s和(b)1 200 s 制備的CuO樣品半電池在1C倍率下循環500圈后的負極片表面形貌圖
3 結 論
基于陽極氧化法及后退火處理在銅箔基底上合成了均勻的氧化銅納米線,該納米線用作鋰離子電池負極材料表現出極好的循環性能,銅箔直接作為集流體,無需任何粘結劑,大大簡化了電池的制備工藝。在1 C的倍率下,陽極氧化1 000 s所對應的樣品表現出594 mAh/g的初始可逆比容量,循環500次的可逆比容量保留率為102.3%。在不同的倍率下,樣品顯示出好的高倍率性能和可逆性能。其優異性能歸因于三維網絡狀納米線結構,交聯的納米線提供了良好的機械性能、較大的比表面和更多的活性位,增加了活性物質與鋰離子的接觸面積,三維網絡結構CuO納米線在鋰離子電池負極材料方面具有良好的應用前景。
