線粒體基因m.3243A>G突變致線粒體糖尿病1例并文獻復習
2020-05-12 07:55:34宋小婷薛存希袁慧娟
河南醫學研究 2020年12期
關鍵詞:糖尿病
宋小婷,薛存希,袁慧娟
(1.錦州醫科大學河南省人民醫院研究生培養基地 內分泌科,河南 鄭州 450003;2.河南省人民醫院 內分泌科,河南 鄭州 450003)
糖尿病是一種以葡萄糖代謝紊亂為特征,廣泛影響全球4億多成年人最常見的代謝性疾病[1]。線粒體基因3243突變糖尿病占一般糖尿患者群的0.5%~2.8%,因該病通常具有母系遺傳和神經性耳聾的臨床特征,故又被母系遺傳糖尿病伴耳聾綜合征 (maternally inherited diabetes and deafness syndrome,MIDD)[2]。線粒體基因突變糖尿病屬于胰島β細胞功能遺傳性缺陷所致特殊類型的糖尿病,是最多見的單基因突變糖尿病,占中國成人糖尿病中的0.6%。絕大多數(85%)線粒體基因突變糖尿病是由線粒體亮氨酸轉運RNA基因[tRNALeu(UUR)]上的線粒體核苷酸序位3243上的A→G(A3243G)突變所致[3-4]。此外,MIDD還可由m.9267G>C、m.1555A>G、m.14530T>C、m.14709T>C、m.3421G>A突變引起[4]。現報道1例典型線粒體糖尿病患者,探討其臨床變現及診治經過,并結合文獻復習總結其臨床特征,以提高臨床醫生對該病的認知,降低誤診漏診率。
1 病例資料
患者,女,31歲,職員。以“口干、多飲、多尿14 a,耳鳴伴聽力下降8 a”為主訴入院。患者14 a前出現口干、多飲、多尿、體質量減輕,于當地醫院確診為2型糖尿病,并給予口服降糖藥二甲雙胍聯合拜糖平治療,血糖控制不佳,空腹血糖10 mmol·L-1。遂改為諾和靈R 30 U皮下注射,每日2次,血糖控制仍差。8 a前出現耳鳴伴聽力下降,飲食睡眠差,體質量減輕7 kg。其母親及外祖母患有糖尿病。為進一步診治,入住河南省人民醫院內分泌科。入院后查體:身高159 cm,體質量60 kg,體質量指數:23.7 kg·m-2,慢性面容,雙眼視力無明顯下降,雙耳聽力下降,雙足均有胼胝體形成,四肢觸覺。位置覺、涼溫覺未見明顯異常,雙側足背動脈搏動無明顯減弱,雙下肢無明顯水腫。常規檢查:腎功能檢查示空腹血糖為20.5 mmol·L-1,尿常規示尿糖(+++),酮體(-),血脂檢查示總膽固醇6.22 mmol·L-1,甘油三酯3.83 mmol·L-1,高密度脂蛋白膽固醇0.95 mmol·L-1,低密度脂蛋白膽固醇3.86 mmol·L-1,彩超提示脂肪肝,血常規、肝功能、電解質、甲狀腺功能、性激素未見明顯異常,心電圖、胸片均未見明顯異常。糖尿病相關檢查:糖化血蛋白10.6 %,尿蛋白檢查示尿微量白蛋白/尿肌酐82.9 mg·g-1,24小時尿蛋白0.21 g,胰島素抗體檢查:抗胰島素抗體(-),抗胰島細胞抗體(-),抗谷氨酸脫氫酶抗體(-)。眼底照相未見明顯異常,四肢肌電圖檢查示雙上肢神經感覺傳導通路異常,聽力閾值測定示神經性耳聾。糖耐量及胰島功能檢查結果見表1。初步診斷:2型糖尿病伴多發癥(神經性耳聾、周圍神經病變、腎臟病變);高脂血癥;脂肪肝。對患者予以糖尿病教育,胰島素泵控制血糖,口服硫辛酸、甲鈷胺、依帕司他膠囊改善神經系統癥狀,厄貝沙坦改善尿蛋白,阿托伐他汀控制血脂,入院治療1周左右,血糖趨于平穩,口干、多飲、多尿癥狀改善,耳鳴、聽力下降癥狀改善不明顯。故重新回顧患者病史,調整診斷思路,行肌酶譜檢測,未見明顯異常,血乳酸檢測:靜息狀態為4.3 mmol·L-1,活動后血乳酸為8.7 mmol·L-1(正常值0.1~2.7 mmol·L-1),行血液和尿液標本的基因檢測,線粒體基因存在m.3243A>G突變(見圖1),故該患者最終診斷:線粒體糖尿病伴多發癥(神經性耳聾、周圍神經病變、腎臟病變);高脂血癥;脂肪肝。調整治療方案:(1)糖尿病教育:不嚴格控制飲食,避免劇烈運動;(2)降糖:賴脯胰島素+諾和靈N;(3)改善線粒體功能、抗氧化:輔酶Q10、硫辛酸膠囊、維生素C;(4)降脂:阿托伐他汀片。患者耳鳴、聽力下降癥狀改善,病情穩定后出院繼續對癥支持治療。

表1 糖耐量試驗及胰島功能檢查結果
注:OGTT—口服葡萄糖耐量試驗。
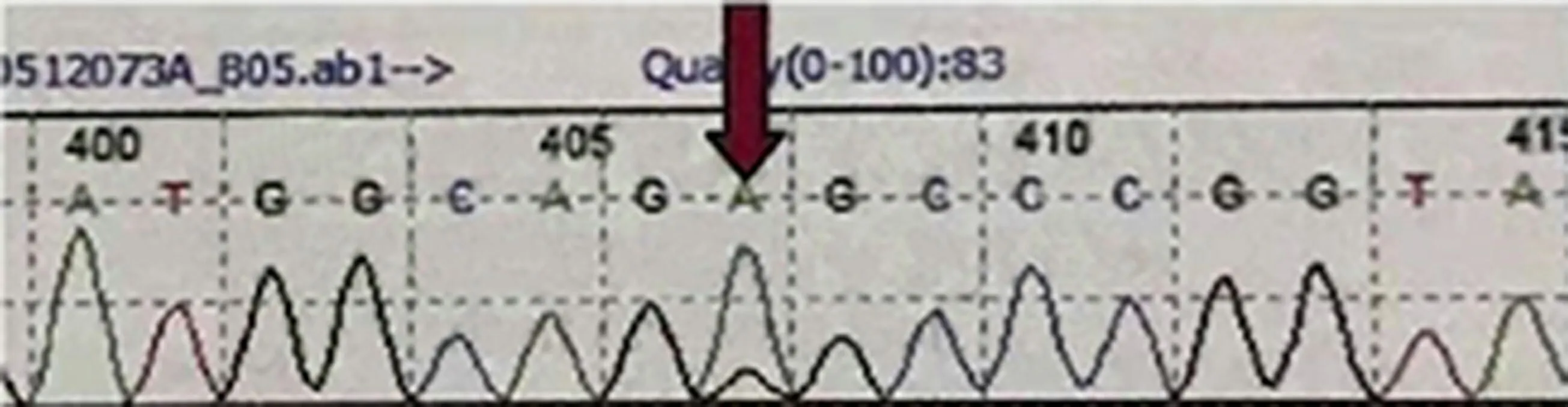
圖1 線粒體基因存在m.3243A>G突變
2 討論
本例患者被誤診為2型糖尿病,有以下臨床特點:(1)青少年起病,病程長,口服降糖藥物控制血糖不佳轉為胰島素治療,病程中胰島β細胞功能進行性減低,近8 a體質量逐漸下降,胰島自身抗體陰性;(2)合并中樞神經病變,神經性耳聾;(3)合并周圍神經病變,四肢肌電圖檢查示雙上肢神經感覺傳導通路異常;(4)尿微量白蛋白/肌酐提示腎臟損害;(5)家系中3代均有糖尿病患者,40歲以上起病,提示母系遺傳;(6)血乳酸水平升高;(7)行血液和尿液標本的基因檢測,線粒體基因存在m.3243A>G突變;(8)患者無明顯胰島素抵抗,無其他胰腺相關疾病,無特殊體征。此外,該患者無高血壓、甲狀腺激素、性腺激素、電解質未見明顯異常;無藥物及其他化學物品接觸史;患者母親孕期無特殊感染史;患者無反復低血糖發作,血胰島素升高不明顯,無明顯視神經萎縮等眼部癥狀,無尿崩癥,多尿癥狀隨血糖控制后緩解,尿比重正常。
線粒體糖尿病患者起病早,多在40歲以前發病,母系遺傳,體型偏瘦,體質量指數低,胰島自身抗體檢測陰性,口服降糖藥物治療不佳,需要胰島素治療,多伴有神經性耳聾,耳聾可發生在糖尿病前或之后,血乳酸水平升高,本例患者的“三多一少”癥狀和2型糖尿病極為相似,容易誤診漏診,故需要和1型、2型及其他特殊類型的糖尿病進行鑒別診斷,可參考目前國際上采用WHO(1999年)的糖尿病病因學分型體系,中華醫學會建議對疑似者首先應行tRNALeu(UUR)A3243G突變檢測[3]。
線粒體氧化磷酸化產生ATP是有核細胞重要的能量來源,線粒體DNA突變引起線粒體功能障礙,進而線粒體無法產生足夠的ATP,會導致多器官功能缺陷,影響對新陳代謝能量需求很高的組織器官,如神經、骨骼肌肉組織、視網膜、腎臟和胰腺[5-6]。線粒體疾病是罕見的,由線粒體DNA或核DNA突變引起的異質性疾病,可表現出多種臨床癥狀。其中線粒體DNA突變引起的線粒體疾病更為常見,比例為20∶1 000 000,為母系遺傳。核DNA突變較少見(2.9∶100 000),并以常染色體顯性或隱性遺傳[5]。臨床上,線粒體DNAm.3243A>G突變可引起線粒體腦肌病伴乳酸性酸中毒和類中風發作、母系遺傳性糖尿病伴耳聾綜合征、肌陣攣性癲癇伴肌肉破碎紅纖維綜合征、Leigh綜合征、Kearns-Sayre綜合征等多種線粒體疾病[7]。
MIDD最為常見的臨床表現為母系遺傳、糖尿病或伴耳聾[3]。(1)母系遺傳:線粒體DNA主要通過母體卵母細胞遺傳,MIDD中的遺傳異常與母體遺傳有關[8]。MIDD的這一特點表明,家系內女性基因突變者的子女均可能遺傳此病變基因而得病,而男性基因突變者的子女不可能得病。故而根據此特點,盡早進行基因檢測,可通過優生優育指導,阻斷此病在子代的延續。(2)神經性耳聾:感音神經性耳聾是線粒體DNA突變的常見表現之一。張博等[9]研究指出由于內耳細胞內突變的線粒體DNA不斷積累,ATP生成不能滿足細胞能量需求,致使神經傳導受阻,聽力逐漸喪失。另一項研究顯示,在A3243G突變的患者中,30%~75%的患者存在感音神經性耳聾[7]。A3243G突變導致聽力受損的臨床特征是雙側對稱性感音性感音神經性耳聾,最初較高頻率聽力受損,隨后較低頻率聽力受損。(3)糖尿病:大多數MIDD患者糖尿病起病隱匿,類似2型糖尿病,此外,高達8%的MIDD病例可表現為1型糖尿病,這取決于胰島功能受損的嚴重程度。在MIDD患者中,由于胰島素敏感性正常,二甲雙胍療效不如2型糖尿病患者,且二甲雙胍治療使乳酸性酸中毒風險增高[10]。(4)其他系統表現:在MIDD的最初研究報道中指出,該病對胰島功能和中樞神經系統造成影響,但在隨后幾年的研究證明,MIDD不僅局限于2個器官,而是線粒體多器官功能紊亂綜合征,除中樞神經系統和內分泌器官受累外,MIDD還可表現為周圍神經系統、前庭功能、眼睛、心臟、胃腸道和腎臟等多系統臟器受到影響[11]。
鑒于線粒體糖尿病的臨床特點,一旦臨床上發現早發糖尿病、體質量減輕、母系遺傳、口服降糖藥效果不佳、較快行胰島素治療或伴神經性耳聾的患者,應及時行基因檢測,盡早明確診斷,調整治療方案,改善患者生活質量,指導優生優育,降低誤診率。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年3期)2021-08-22 06:49:56
中老年保健(2021年11期)2021-08-22 03:15:16
中國生殖健康(2020年2期)2021-01-18 02:51:44
中國生殖健康(2018年2期)2018-11-06 07:11:04
基層中醫藥(2018年2期)2018-05-31 08:45:04
