酚二磺酸光度法測定硝酸鹽氮方法誤差分析
2020-05-15 02:59:40
環境科學導刊 2020年2期
(紅河州環境監測站,云南 蒙自 661100)
在硝酸鹽氮的諸多測定方法中,酚二磺酸分光光度法由于測量范圍較寬、顯色穩定、測量儀器易得成為測定硝酸鹽氮的常用方法。但是,在實際樣品分析中往往會出現加標回收率偏高的現象,究其原因,在酚二磺酸分析方法中,校準曲線的測定條件與樣品的測定條件不一致,不知這是否是引起加標回收率偏高的原因。為此,在按照分析方法[1]測定標準曲線的同時,也進行工作曲線的測定,并將標準曲線與工作曲線進行比較。經統計檢驗,標準曲線與工作曲線之間存在著系統誤差。
1 校準曲線的繪制
1.1 測定條件的選擇
酚二磺酸法的測定原理是硝酸鹽氮在無水情況下與酚二磺酸反應生成硝基二磺酸酚,在堿性溶液中生成黃色化合物,進行測定。在分析過程中,首先要將水樣置于蒸發皿內在水浴上蒸干,為避免蒸發過程中硝酸鹽氮的損失,需用氫氧化鈉溶液調節樣品pH值至8,樣品的pH值過高或過低均會對測定結果產生影響。在酸度計上測得50mL硝酸鹽標準溶液中加入1滴0.1mol/L氫氧化鈉溶液的pH為10左右,溶液pH值過高。因此,在樣品測定的實際操作中,將氫氧化鈉溶液的濃度改為0.025mol/L,滴加2滴,即可達到樣品pH值8的要求。實驗證明瓷蒸發皿表面的光潔度對測定結果也會產生影響,瓷蒸發皿長期在偏堿性條件下使用表面會受損,容易對硝酸鹽吸附而影響測定結果。因此,在樣品測定中應選用表面光潔的瓷蒸發皿以減少瓷蒸發皿對被測物質的吸附。
1.2 校準曲線的測定
標準曲線的測定:吸取50.00mL硝酸鹽貯備液置于蒸發皿內,加入2滴0.025mol/L氫氧化鈉溶液,在水浴上蒸發至干。加入2.0mL酚二磺酸,用玻棒研磨蒸發皿內壁,使殘渣與試劑充分接觸,放置片刻,再重復研磨一次,放置10min,加入少量水,將溶液移入500mL溶量瓶中,稀釋至標線,混勻,得到0.010mg/mL標準使用液。按標準系列吸取適量的標準使用液于50mL比色管中,加水至約40mL,加3mL氨水使成堿性,稀釋至標線,混勻。在波長400nm處以水為參比,以10mm比色皿光程測量吸光度。
工作曲線的測定:將硝酸鹽貯備液用純水稀釋成0.010mg/mL硝酸鹽使用液,按標準系列吸取適量的使用液于各蒸發皿中,加水至50mL,加入2滴0.025mol/L氫氧化鈉溶液,在水浴上蒸發至干。加入1.0mL酚二磺酸,用玻棒充分研磨蒸發皿內壁,放置片刻,再重復研磨一次,放置10min,將溶液移入50mL比色管中,加入3mL氨水顯色,定容,在波長400nm處比色皿光程10mm以水為參比測量吸光度。
在制備一份硝酸鹽標準使用液的同時,測定一條工作曲線。標準曲線、工作曲線各測定4次。
1.3 測定結果
由測得的吸光度值減去零濃度管的吸光度值,繪制校準曲線。結果見表1。
2 方法誤差分析
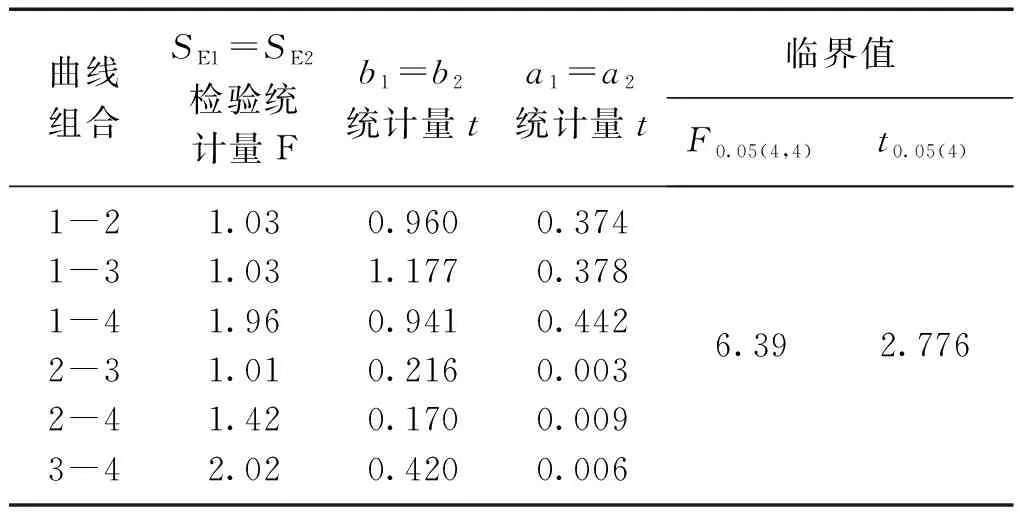
方法誤差分析的步驟:為了避免偶然性,在誤差分析過程中對多條曲線以及其平均水平進行了比較。首先對各校準曲線回歸直線的線性密切程度(相關系數)和截距(a=0)進行統計檢驗,以考查校準曲線的繪制是否達到要求。若校準曲線繪制合格,再進行任意兩條標準曲線之間的比較,如果檢驗結果無顯著差異,將四條標準曲線進行合并得到一條平均標準曲線。對工作曲線也進行同樣的檢驗,無顯著差異,再進行四條工作曲線的合并。然后對同步測定的標準曲線與工作曲線及合并后的標準曲線與工作曲線進行比較,看其是否存在差異。
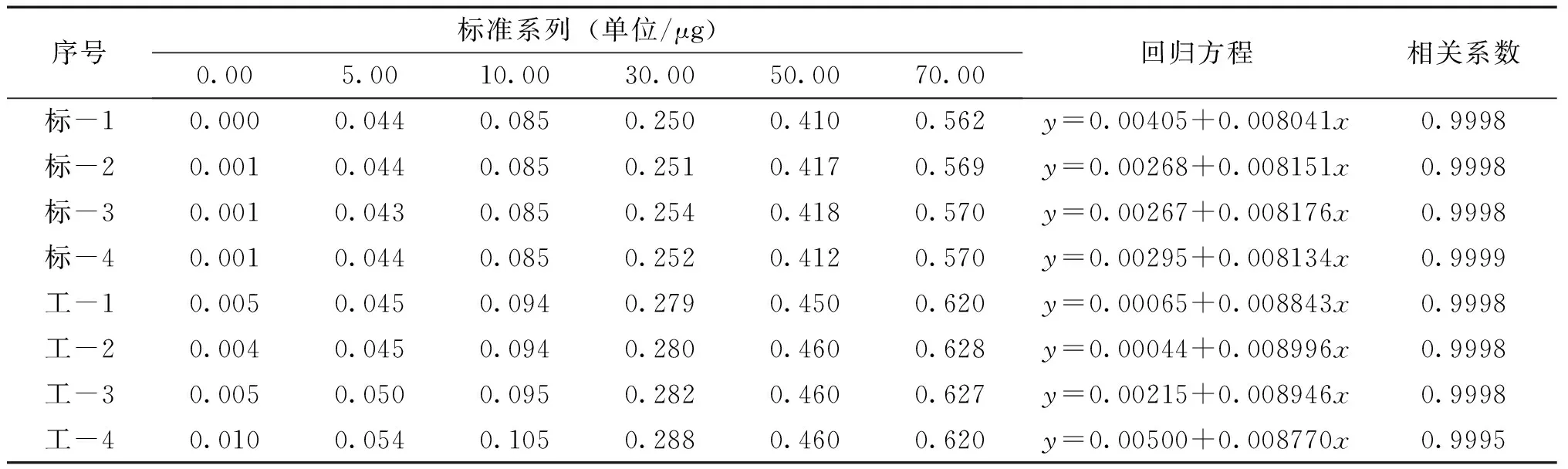
表1 校準曲線測定值及回歸方程
2.1 校準曲線的線性和截距檢驗
對于以4~6個濃度單位所獲得的測量信號值繪制的校準曲線,分光光度法一般要求其相關系數︱r︱≥0.9990[1],從表1可以看出八條校準曲線的精密度都達得到要求。
校準曲線截距a=0的統計檢驗,統計量t按下式計算。當a與0有顯著性差異時,說明校準曲線的回歸方程計算結果準確度不高。

表2 校準曲線截距a=0檢驗結果
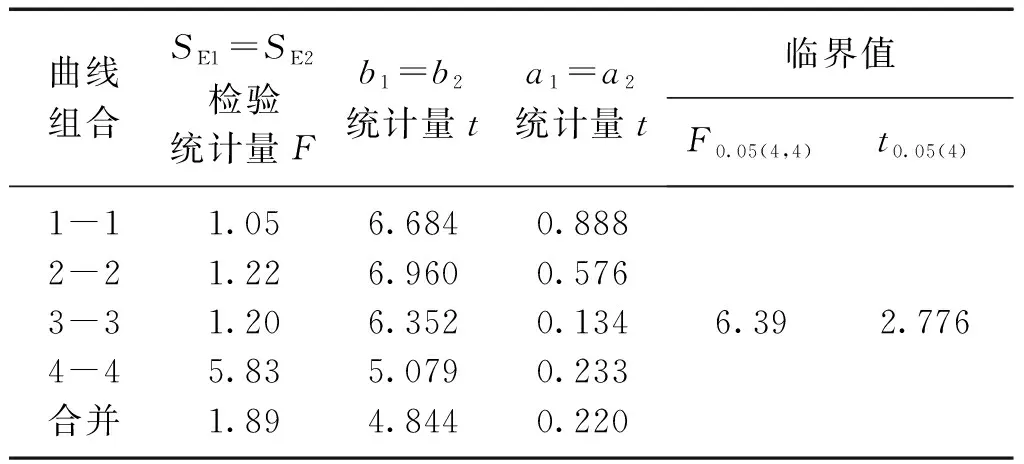
檢驗結果,t 對不同時間測定的兩條工作曲線的差異性進行檢驗,統計量的計算與標準曲線相同。 對同步測定的標準曲線與工作曲線以及合并后得到的平均標準曲線與平均工作曲線進行比較,看其是否存在顯著差異。檢驗方法與標準曲線的差異性檢驗相同。 表3 標準曲線各統計量計算結果 檢驗結果,F 表4 工作曲線各統計量計算結果 檢驗結果,F 表5 標準曲線與工作曲線各統計量計算結果 檢驗結果,F 標準曲線和工作曲線各濃度點的測量信號值之間的差異性檢驗。對于系列相同的兩組測定值,如果其測定的隨機誤差和兩組測定值之間的系統誤差都很小,則以橫坐標(x)表示其中一組測定值,以縱坐標(y)表示另一組測定值所得到的點應分布在通過原點且斜率為1的直線兩側。這些點的回歸方程的截距和回歸系數反映了這兩組測定值的系統誤差。以橫坐標表示工作曲線各濃度點的測量信號平均值,以縱坐標表示標準曲線各濃度點的測量信號平均值,建立回歸方程y=0.0013+0.9136x,相關系數r=0.9999,對回歸直線的截距和回歸系數進行檢驗。 截距a=0的檢驗,計算統計量t=0.568,t 回歸系數b=1的檢驗: ︱t︱>t0.05(4)(2.776),回歸直斜率不為1,兩組測量值間確實存在著系統誤差。 通過標準曲線與標準曲線、工作曲線與工作曲線、標準曲線與工作曲線之間的差異性檢驗,標準曲線之間、工作曲線之間無顯著差異;標準曲線與工作曲線之間存在顯著差異,只是在原點附近的區域內差異較小。以標準曲線進行計算,樣品分析結果將偏高8%左右,酚二磺酸光度法存在正的系統誤差。引起系統誤差的原因是由于方法所采用的校準曲線即標準曲線在繪制過程中,制備標準使用液時硝基化率偏低,使得標準曲線斜率偏低。因此,要克服方法中存在的系統誤差,在樣品分析中應采用與樣品分析步驟相一致的工作曲線作為校準曲線進行分析結果的計算。2.2 標準曲線的差異性檢驗
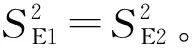
2.3 工作曲線的差異性檢驗
2.4 標準曲線與工作曲線的差異性檢驗
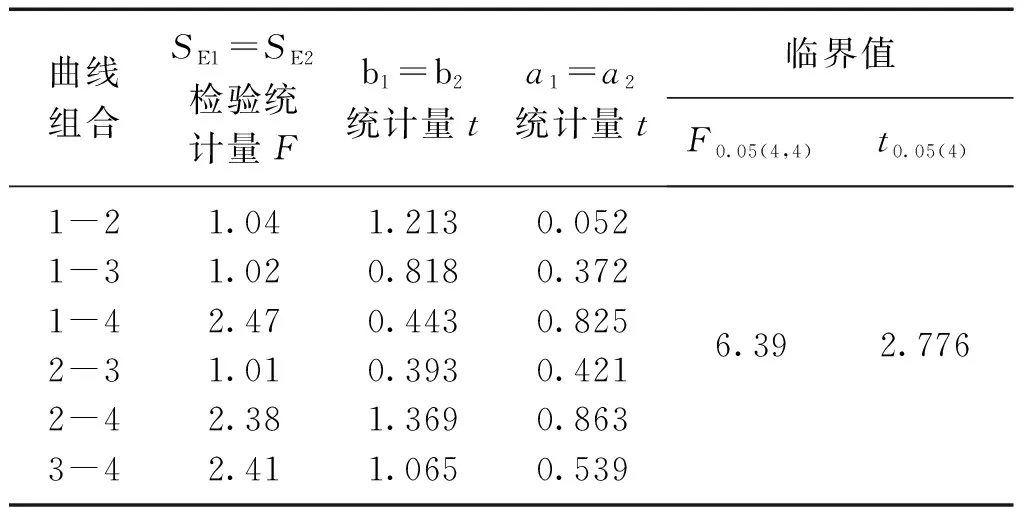
3 結論
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12音樂探索(2022年2期)2022-05-30 21:01:37小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00當代陜西(2019年8期)2019-05-09 02:22:48動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10中國特種設備安全(2018年11期)2019-01-08 02:08:32小學科學(學生版)(2018年7期)2018-08-13 09:33:04家庭影院技術(2018年4期)2018-05-09 07:07:52專用汽車(2016年4期)2016-03-01 04:13:43質量與標準化(2015年9期)2015-12-31 11:41:40
