延胡索中叔胺堿和季銨堿類組分的高效分離及鎮痛活性鑒定
2020-05-16 04:14:10高振華王超然郭秀潔王紀霞郭志謀王聯芝
天然產物研究與開發 2020年3期
高振華,王超然,郭秀潔,王紀霞,郭志謀,王聯芝
1湖北民族大學,恩施 44500;2中國科學院大連化學物理研究所,大連 116023;3中科院大化所中國醫藥城生物醫藥創新研究院,泰州 225300
延胡索為罌粟科紫堇屬植物延胡索CorydalisyanhusuoW.T.Wang的干燥塊莖,別名元胡、延胡、玄胡索、元胡索等,是傳統的“浙八味”之一[1,2]。作為一味應用歷史悠久的活血、行氣、止痛中藥,其療效一直為歷代醫家推崇,目前也仍廣泛應用于治療心律失常、氣虛瘀滯、胸痹心痛、脘腹疼痛、產后瘀滯腹痛、跌打損傷等,是中藥復方制劑中的常用藥[3,4]。
現代研究表明,延胡索中主要活性成分為生物堿,其中以叔胺型的原小檗堿類和季銨型的小檗堿類含量最高,占比可分別達到0.65%和0.3%左右[5-7]。這兩類成分骨架結構非常類似,但生物學效應大不相同。藥理學研究表明,延胡索的鎮痛、鎮靜、催眠和安定等作用主要與水溶性極差的叔胺型生物堿相關[8,9],其中以乙素、丑素最強,甲素次之,雖然都比嗎啡弱,但其作用受體為多巴胺受體,沒有成癮性缺陷,耐藥性也優于嗎啡。相比之下,延胡索中的季銨堿成分,在鎮痛方面發現較少,但在抗心肌缺血、較少心肌耗氧等方面具有更顯著的作用[10-12]。
延胡索中叔胺堿和季銨堿的不同作用靶點和功效,提示了分別以叔胺堿和季銨堿類組分開發活性部位制劑將使藥效作用機制更加清晰,在治療具體疾病時更有針對性,具有中藥5類新藥的開發前景,而目前對延胡索作用機制和制劑產品的研究仍主要以總堿為主[13,14],成分較復雜,往往難以有效闡明療效物質基礎。也有部分研究工作采用樹脂層析或酸堿萃取的方式,得到了延胡索叔胺堿或季銨堿的組分,但總體工藝穩定性較差,酸堿廢液量大,難以符合現代的環保生產要求,得到的組分純度也不高[15,16]。在前期分離純化延胡索生物堿單體的過程中,我們也發現這兩類成分在色譜填料上的色譜峰形和保留行為具有很大差異,將其采用C18WCX實現類組分分離后,分別采用C18和C18SCX來制備叔胺堿和季銨堿單體,可以使兩類成分在不同的分離方法中獲得良好的色譜峰形和載樣量,從而降低純化制備的難度[17]。因此,發展高效的延胡索生物堿類組分制備方法,不僅對延胡索類組分制劑的開發具有重要作用,對生物堿單體的制備,深入研究延胡索的組成成分也能帶來很大的便利。
為克服使用C18WCX填料進行類組分制備過程中載樣量不易控制,填料比較小眾不易產業化等缺點,本文采用普通C18填料,利用延胡索叔胺堿和季銨堿在不同pH值條件下電荷性質不同的特點,發展了一種更為簡便易行的延胡索類組分高效制備方法,并對得到的兩類組分進行了生物堿含量測定和多巴胺D2受體拮抗活性的驗證,結果顯示,兩類成分在新方法中得到了有效分離,且類組分的生物堿含量以已知對照品計分別達到了84.0%和60.7%,符合有效部位中藥制劑的開發要求,延胡索提取物的多巴胺D2受體拮抗活性也集中在了叔胺堿組分中,可為相應的類組分新藥制劑開發奠定基礎。
1 材料與方法
1.1 儀器、試劑與材料
實驗儀器:Alliance e2695高效液相色譜儀(美國Waters公司)包括自動進樣器,2695型分離單元,柱溫箱系統,2996型光電二極管陣列檢測器和 Empower 色譜工作站。YJD20D-GL十功能自動煎藥機(北京東華原醫療設備有限責任公司)、LX-0400型20 L旋轉蒸發儀(上海申勝生物技術有限公司)、CeraMem0100-015型500 nm陶瓷膜(廈門福美科技有限公司)、DAC-50動態軸向壓縮柱(江蘇漢邦科技有限公司)、PUERLAB Chorus實驗室純水機(英國ELGA公司)、FLIPR Tetra 高通量實時熒光檢測分析系統(美國Molecular Devices公司)、SCIENTZ-10N冷凍干燥機(寧波新芝生物科技股份有限公司)。
使用的色譜柱:Kromasil 100-5-C18柱(4.6×150 mm,5 μm,Kromasil,瑞典);C18柱(4.6×250 mm,10 μm,大連思譜,大連)。
延胡索藥材樣品(產自浙江金華)。乙腈、乙醇、醋酸、醋酸銨均為分析純(購自上海國藥集團化學試劑有限公司)、多巴胺、氟哌啶醇(購自梯希愛上海仁成工業發展有限公司)、制備乙醇(購自上海星可高純溶劑有限公司)。所使用的對照品(1∶1,3-甲基去氫紫堇達明堿;2:去氫紫堇球堿;3:巴馬汀;4:去氫紫堇堿;5:海罌粟堿;6:延胡索乙素;7:延胡索甲素.)均為實驗室自制,HPLC檢測純度為95%以上。制備實驗所使用的C18填料購自大連思譜精工有限公司,粒徑10 μm,孔徑10 nm。固相萃取實驗所使用的C18CEX填料購自大連思譜精工有限公司,粒徑40 μm,孔徑10 nm。
1.2 實驗過程
1.2.1 延胡索生物堿提取物制備
取延胡索干燥樣品500 g,放入十功能自動煎藥機,第一次提取加入70%乙醇水溶液5 L,提取溫度80 ℃,沸騰后煎煮60 min,第二次提取加入70%乙醇水溶液5 L,溫度80 ℃,沸騰后煎煮40 min,合并兩次提取液。用20 L旋轉蒸發儀進行濃縮去除乙醇至1.6 L后,使用500 nm陶瓷膜進行澄清過濾,收集透過液,至循環端液體體積降低至200~300 mL后,加0.8 L純化水進行洗濾,合并膜透過液,得到延胡索澄清透過液1.80 L,旋蒸濃縮至180 mL,取2.0 mL樣品凍干得固體0.245 g,測得其固含量為122.5 g/L,總提物為22.05 g。
1.2.2 延胡索生物堿提取物樣品分析和類組分制備方法開發
樣品分析:Kromasil 100-5-C18(5 μm,4.6×150 mm)色譜柱,流動相A相為乙腈,B相為0.1%磷酸(三乙胺調節pH值至6.0)或0.1 mol/L醋酸銨(加醋酸調節pH值為6.0、6.5、6.8),C相為純水,梯度洗脫條件:0~20 min,23% A;20~30 min,23%→55% A;30~50 min,55% A;B相固定20%。檢測波長280 nm,流速1 mL/min,進樣量10 μL,柱溫30 ℃。
類組分制備方法開發:C18(10 μm,4.6×250 mm)色譜柱,流動相A為乙醇,B為20 mmol醋酸銨(pH值為6.8),梯度洗脫條件:0~20 min,23% A;20~30 min,23%←55% A;30~50 min,55% A。臺階等度洗脫條件:0~30 min,23%A;30~30.1 min,23%~55%A;30.1~50 min,55%A。檢測波長280 nm,流速0.6 mL/min,進樣量10 μL,柱溫30 ℃。
1.2.3 延胡索類組分制備
制備柱為DAC-50 mm動態軸向壓縮柱,填裝C18(10 μm,大連思譜,大連)填料300 g,流動相A為乙醇,B為20 mmol醋酸銨(pH值為6.8),梯度洗脫條件:0~30 min,23%A;30~30.1 min,23%→55%A;30.1~50 min,55%A。檢測波長280 nm,流速70 mL/min。
1.2.4 延胡索類組分含量測定
對照品溶液的配制:使用十萬分之一天平,準確稱取延胡索乙素、延胡索甲素和海罌粟堿各10 mg,使用含0.1%甲酸的50%甲醇進行溶解,配制成一系列梯度的叔胺堿混合對照品溶液,另稱取去氫紫堇堿、巴馬汀、去氫紫堇球堿、1,3-甲基去氫紫堇達明堿各10 mg,使用含0.1%甲酸的50%甲醇進行溶解,配制成一系列梯度的季銨堿混合對照品溶液。以混合對照品的濃度為橫坐標、峰面積為縱坐標建立標準曲線。結果表明,叔胺堿混合標準品在3.2~2 000 mg/mL范圍內線性關系良好,線性相關系數(r2)均大于0.999 7,季銨堿混合標準品在40~1 000 mg/mL范圍內線性關系良好,線性相關系數(r2)均大于0.999 9。
延胡索類組分供試品制備:稱取類組分制備得到的叔胺堿和季銨堿類組分凍干粉末各10 mg,使用50%甲醇水溶液超聲溶解后于10 mL容量瓶中定容,配置成1 mg/mL的叔胺堿及季銨堿類組分供試品溶液。
1.2.5 鎮痛活性測試實驗條件
將延胡索提取物及類組分樣品取樣,在表達多巴胺D2受體的細胞人胚腎293T細胞(HEK293T)上,采用FLIPR(Fluorometric Imaging Plate Reader,Molecular Device Corp)進行活性篩選,樣品終濃度為50 μg/mL。以每孔80 000個細胞接種于用poly-D-lysine涂層的96孔細胞培養板中,24 h后除去培養基并于每孔中加入100 μL熒光染料溶液(calcium-6)在37 °C下恒溫1 h時,除去染料溶液并于每孔加入100 μL 0.5% amaranth,將待測樣品用二甲基亞砜(DMSO)溶解后置于96孔板中,采用FLIPR進行自動加樣與細胞培養板中孵育10 min后在520和488 nm波長下進行熒光檢測從而對細胞內的Ca2+濃度進行監測。
2 結果與討論
2.1 延胡索提取物樣品分析
參考文獻方法[18,19],我們首先以乙腈和磷酸三乙胺緩沖鹽(pH 6.0)為流動相對延胡索提取物進行了分析。結果如圖2a所示,延胡索主要生物堿成分都得到了較好的分離,20 min以后峰色譜峰峰形良好,但5~20 min區域的色譜峰均較拖尾。通過DAD檢測器的色譜峰紫外光譜圖分析可知(如圖3所示),峰1~6均具有季銨型小檗堿類生物堿的特征吸收,而峰8~12均為叔胺型原小檗堿類生物堿的特征吸收,峰7為海罌粟堿為阿樸啡類生物堿的特征吸收。通過與實驗室前期自制的標品對比,2、3、4、6號峰分別為1,3-甲基去氫紫堇達明堿、去氫紫堇球堿、巴馬汀、去氫紫堇堿(均為季銨堿),峰9、12分別為延胡索乙素、延胡索甲素,峰7為海罌粟堿(均為叔胺堿),也證實兩類成分在接近中性的流動相體系下,色譜保留時間存在較大差異,能夠實現兩類組分的良好分離。在該pH條件下,堿性較弱的叔胺堿電離已經得到大部分抑制,主要呈分子態,保留較強,色譜峰形也較好,而季銨堿永久帶電荷,呈離子狀態,因而保留總體弱于叔胺堿。
磷酸三乙胺不易揮發,且在后續工業生產中無法用膜分離回收套用,為便于后續的液質聯用分析和工業化制備應用,我們嘗試用中性的醋酸銨代替磷酸三乙胺。如圖2b所示,0.1 mol/L 醋酸銨(pH 6.8)條件下,叔胺堿與季銨堿同樣可以實現良好的分離,且具有鎮痛活性的海罌粟堿與叔胺堿更為接近,可將其歸入叔胺堿類組分。但海罌粟堿與8號峰的保留過于接近,不利于含量分析,經pH微調,發現pH為6.5時,7號峰海罌粟堿能與周邊峰得到更好的分離,故選擇pH6.5為分析條件,而在類組分制備時可選擇pH6.8。
2.2 延胡索類組分制備方法開發與組分制備
在傳統方法中,常使用大孔吸附樹脂對延胡索生物堿成分純化分離,但是大孔吸附樹脂對溶劑需求量大,樣品分離過程緩慢,且大孔吸附樹脂再生復雜,不適合大量樣品的分離制備[20]。制備色譜通常使用10 μm以上的填料,且優先使用甲醇、乙醇等易于納濾回收且價格相對低廉的有機溶劑,以節約溶劑的使用成本。本實驗采用了10 μm的C18填料,先使用4.6×250 mm的預裝柱上進行溶劑和梯度方法考察。通過實驗發現,在乙醇/醋酸銨(pH 6.8)體系下,采用相同的梯度條件,延胡索提取物中的季銨堿雖然峰形更為拖尾,但與叔胺堿的類分離由于叔胺堿的保留增強,類分離效果更優(如圖4a所示),這也可能是制備填料與分析柱填料選擇性略有差異導致。為便于生產制備,將梯度條件簡化為23%乙醇和55%乙醇兩個臺階梯度,分別用于洗脫季銨堿和叔胺堿。考慮到圖4a中20 min 23%的乙醇還不能把季銨洗脫完全,因此把季銨堿的洗脫時間延長到30 min,結果如圖4b所示,叔胺堿和季銨堿的分離度進一步擴大。
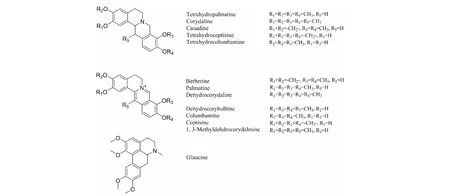
圖1 延胡索中主要的叔胺堿和季銨堿Fig.1 Structures of main tertiary and quaternary alkaloids in Rhizoma Corydalis
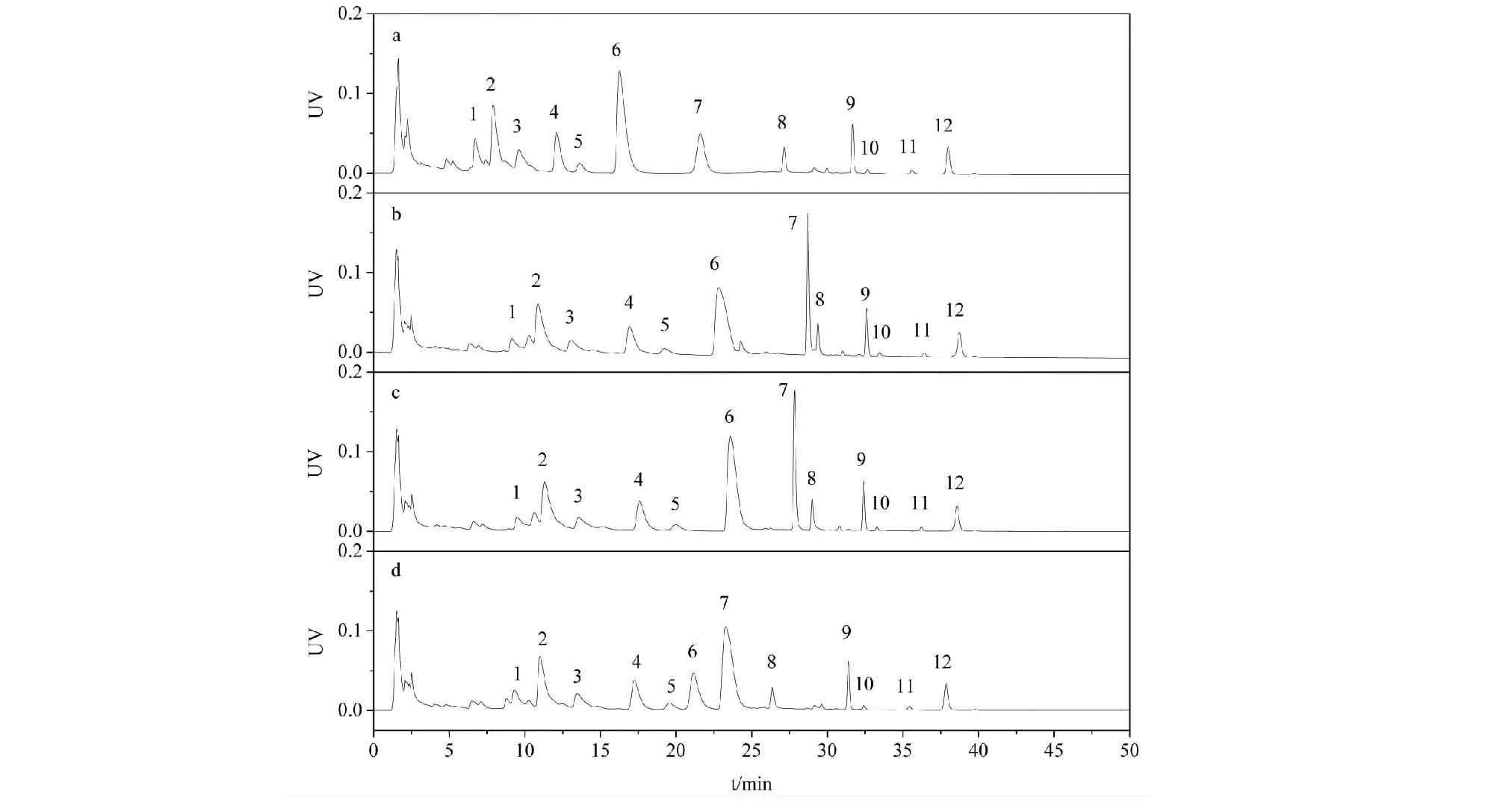
圖2 延胡索提取物在不同緩沖鹽條件下的分離譜圖Fig.2 Separation chromatogram of Rhizoma Corydalis extracts with different buffer salt 注:流動相添加劑:(a)0.1%磷酸三乙胺;(b~d)0.1 mol/L醋酸銨,pH值分別為6.8、6.5、6.0。洗脫梯度(a):0~20 min,23% A;20~30 min,23%→55% A;30~50 min,55% A;洗脫梯度(b~d):0~20 min,23% A;20~30 min,23%→55% A;30~50 min,55% A,緩沖鹽相固定20%。色譜峰:叔胺堿:7.海罌粟堿;9.延胡索乙素;12.延胡索甲素;季銨堿:2.1,3-甲基去氫紫堇達明堿;3.去氫紫堇球堿;4.巴馬汀;6.去氫紫堇堿。Note:Additives:(a) 0.1% triethylammonium phosphate;(b-d) 0.1 mol/L ammonium acetate with pH at 6.8,6.5,6.0 respectively.gradient(a):0-20 min,23%A;20-30 min,23%→55%A;30-50 min,55%A.gradient(b-d):0-20 min,23%A;20-30 min,23%→55%A;30-50 min,55%A,buffer salt phase fixed at 20%.Peaks:Tertiary alkaloids:7.Glaucine;9.Tetrahydropalmatine;12.Corydaline;Quaternary alkaloids:2.1,3-Methyldehdrocorydalmine;3.Dehydrocorybulbine;4.Palmatine;6.Dehydrocorydaline.
圖3 延胡索提取物主要色譜峰的紫外吸收譜圖Fig.3 Ultraviolet spectra of main chromatographic peaks of Corydalis extracts
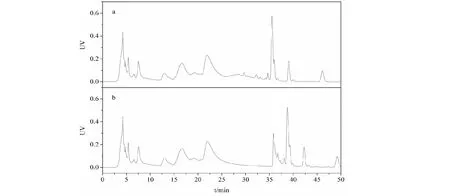
圖4 乙醇/醋酸銨體系梯度及臺階等度色譜圖Fig.4 Gradient and step equivalent chromatography of ethanol/ammonium acetate system
在選定的流動相條件下,將方法轉移至裝填有300 g相同C18填料的DAC-50 mm制備柱上進行放大。經簡單摸索,樣品上樣量為80 mL(含固體9.8 g,相當于填料量3.3%)時的制備譜圖如圖5所示:
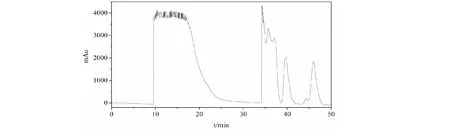
圖5 延胡索類組分制備液相色譜圖Fig.5 Preparative chromatogram of Rhizoma Corydalis class separation
分別收集10~20 min和34~50 min的餾分,進行HPLC分析,分析結果如圖6所示,可以看到兩類組分實現了良好的分離,兩類生物堿之間無成分交叉。從制備譜圖和分析結果看,樣品載樣量還有比較大的提升空間,但本次實驗因為提取物樣品量有限,上樣量先增加到了填料量的3.3%,在工業化生產過程中樣品載樣量可以繼續增大,適用于大規模樣品的生產制備。
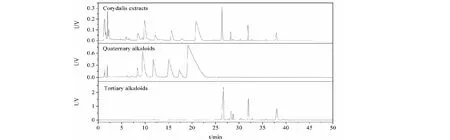
圖6 制備餾分分析色譜圖Fig.6 Analytical chromatograms of preparative fractions
2.3 延胡索類組分的后處理與含量測定
上述制備方法制得的延胡索類組分含有醋酸銨緩沖鹽,在工業生產中,可使用納濾膜濃縮,實現溶劑回收套用,但本次小試的樣品量較少,采用固相萃取(SPE)的方式進行同步脫鹽和濃縮。實驗使用一種耐純水的親水性反相填料C18CEX作為SPE固定相,將類組分制備所得餾分經旋蒸濃縮除去乙醇后,分別上樣至兩個活化平衡好的20 g裝SPE柱,水洗3倍柱體積脫鹽后,甲醇洗脫,旋蒸,凍干得到固體粉末,其中季銨堿固體粉末0.68 g ,叔胺堿固體粉末1.00 g。
按前述分析方法,以實驗室自制的7個已知成分為對照品,建立標準曲線,對所制得的類組分進行含量測定。計算結果顯示,在叔胺堿類組分中延胡索乙素含量21.0%,延胡索甲素含量21.0%,海罌粟堿含量42.0%,以該3個主要已知叔胺堿計,叔胺型類組分的生物堿含量達84.0%,而在季銨堿類組分中去氫紫堇堿含量37.0%,巴馬汀含量7.7%,去氫紫堇球堿含量5.5%,1,3-甲基去氫紫堇達明堿含量10.5%,以這4種已知對照品計,該類組分種含已知成分的含量達60.7%,兩個類組分的已知成分含量總和均超過50%,符合中藥有效部位制劑的已知成分含量大于50%的要求。
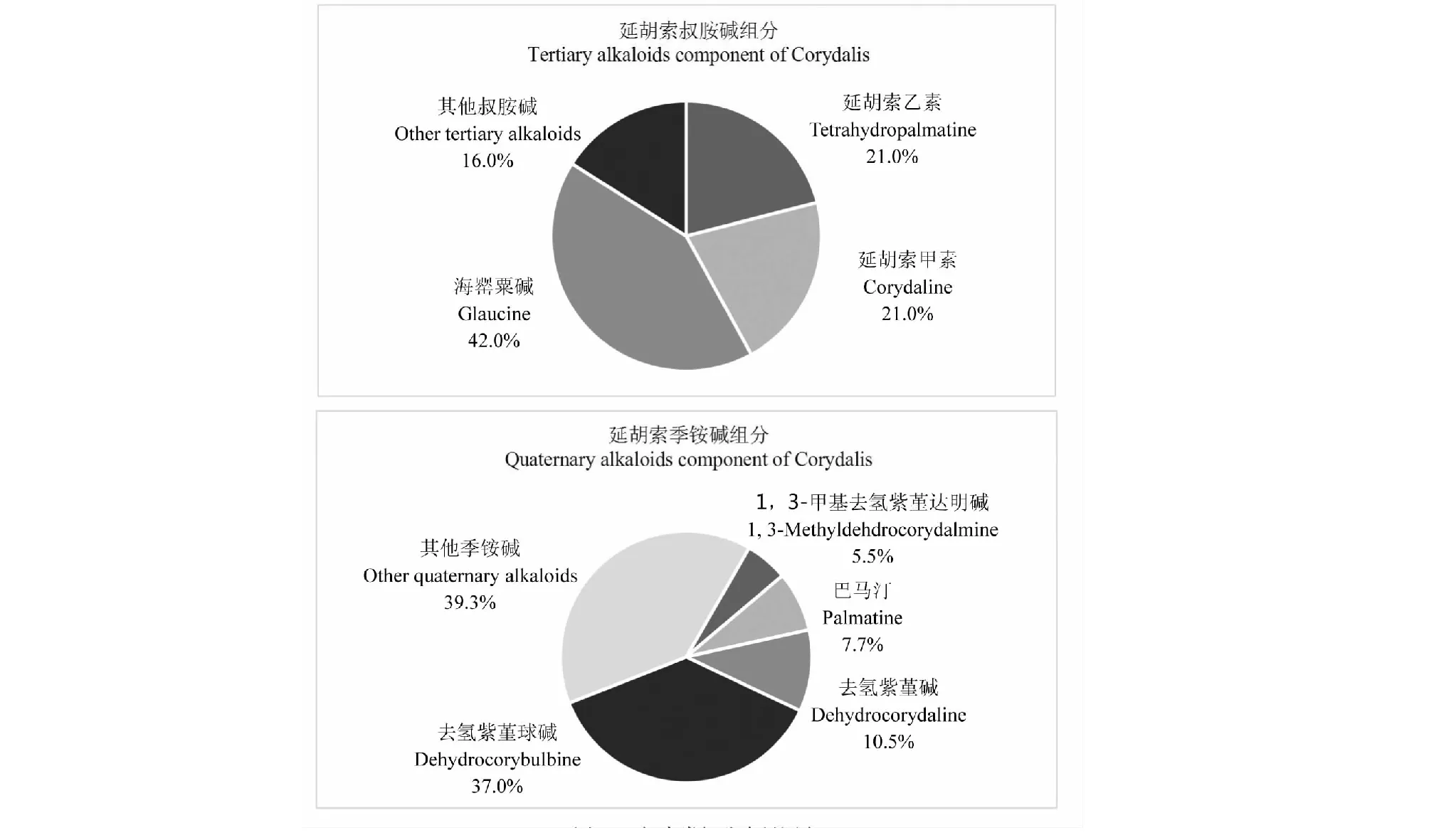
圖7 延胡索類組分含量測定Fig.7 Determination of class components in Rhizoma Corydalis
2.4 延胡索類組分化合物在D2受體上的拮抗作用測試
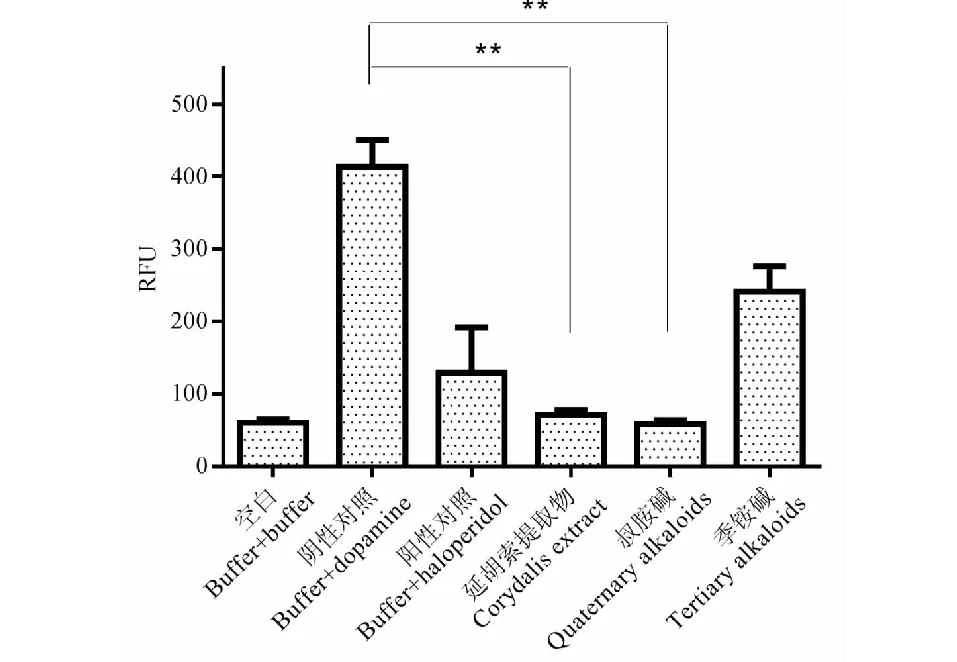
圖8 延胡索組分D2受體拮抗活性測試Fig.8 Antagonist activity of Rhizoma Corydalis fractions on D2 receptor注:與陰性對照組(buffer+dopamine)比較,**P < 0.01。Note:Compared with negative control group (buffer+dopamine),**P < 0.01.
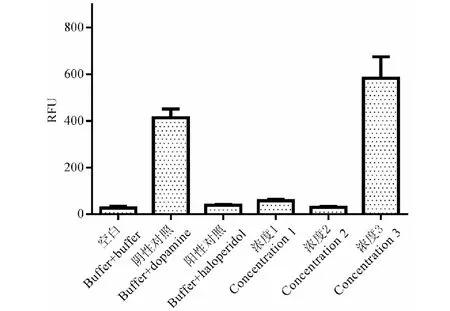
圖9 不同濃度叔胺堿拮抗活性Fig.9 Antagonistic activity of different concentrations of tertiary alkaloid注:濃度1~3分別為50.00、1.85、0.07 μg/mL。Note:Concentration 1-3 were 50.00,1.85,0.07 μg/mL,respectively.
現代藥理學研究表明,延胡索的鎮痛、安定作用是通過多巴胺受體系統發揮作用的,尤其是對多巴胺D2受體的拮抗作用[21]。將制備的季銨堿和叔胺堿類組分與延胡索提取物在Fliper實驗平臺上進行了多巴胺D2拮抗作用的活性對比,結果如圖8所示,延胡索提取物對多巴胺D2受體有顯著拮抗作用,其作用強度與陰性對照組(dopamine)相比具有顯著性差異,且大于陽性對照氟哌啶醇(haloperidol),制備成類組分后,季銨堿類組分的拮抗活性較弱,而叔胺堿的活性強于提取物,這也證明了兩類組分在藥理活性上的差異。叔胺堿組分在不同濃度下都具有拮抗活性,如圖9所示,對叔胺堿樣品進行稀釋并測定其拮抗活性,結果發現濃度降低至1.85 μg/mL時,叔胺堿組分達到最低抑制濃度,拮抗活性明顯,繼續降低濃度直至0.07 μg/mL時,拮抗活性明顯降低,證明了叔胺堿組分的拮抗活性具有劑量依賴性,這些結果為更有治療針對性的有效部位制劑開發提供了科學依據。
3 結論
延胡索中叔胺堿和季銨堿兩類生物堿在電離狀態下保留接近難以區分,但隨著流動相pH值升高,叔胺堿的電離更易受到抑制,保留時間顯著增強,能與季銨堿組分實現有效分離,從而開發了一種簡便的類組分分離方法。通過分離制備得到了兩類成分區分明晰、含量較高,且具有不同藥理活性的生物堿類組分,通過多巴胺D2受體的拮抗活性測試,也進一步證實延胡索的鎮痛相關活性物質主要集中在叔胺堿類組分。這些結果為延胡索生物堿化學成分研究和具有針對性治療作用的有效部位制劑開發奠定了實驗和理論基礎。
