
基于XG-B00ST和多數據源的藥物重定位預測
2020-05-25 02:30:57李苗苗
軟件導刊 2020年2期
李苗苗
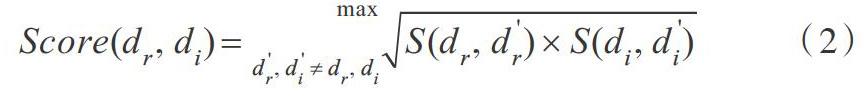

摘 要:新藥物研發時間長、成本高,但成功率低,為了提高收益比,藥物重定位即舊藥新用受到了廣泛關注。從臨床和實驗角度鑒定藥物的新用途需要耗費大量人力和物力,從計算角度預測藥物新用途成為研究熱點;并且,隨著藥物和疾病相關的大量多層次組學數據積累,通過挖掘藥物相關數據鑒定藥物新用途成為可能。重點挖掘藥物化學結構、藥理性質、藥物靶蛋白功能、疾病表型等數據得到相應特征,并將這些藥物疾病特征進行整合,再將特征輸入XG-BOOST模型進行預測。實驗結果表明,該方法準確率達87.9%,較邏輯回歸、隨機森林具有更高的預測精度。
關鍵詞:藥物重定位;XG-BOOST模型;預測精度
DOI:10. 11907/rjdk. 191526 開放科學(資源服務)標識碼(OSID):
中圖分類號:TP301文獻標識碼:A 文章編號:1672-7800(2020)002-0110-04
英標:Drug Reposition Prediction Based on XG-BOOST and Multi-source Data
英作:LI Miao-miao
英單:(Business School, University of Shanghai for Science and Technology, Shanghai 200090, China)
Abstract: The development of new drugs is long and costly, but the success rate is low. Therefore, in order to improve the yield, drug relocation, that is, the new use of old drugs has received extensive attention. The clinical and experimental identification of new uses of drugs requires a lot of manpower and material resources, and predicting the new use of drugs from a computational perspective has become a research hotspot in recent years. On the other hand, in recent years, the rapid accumulation of a large number of multi-level omics data related to drug-related and disease has made it possible to identify new drug uses by mining drug-related data. In this paper, the characteristics of the chemical structure, pharmacological properties, drug target protein function, disease phenotype, etc. of the drug were obtained, and the characteristics of these drugs were integrated. Finally, the feature is input into the XG-BOOST model for prediction. The experimental results show that our method has higher prediction accuracy than logistic regression and random forest.
Key Words: drug reposition;XG-BOOST model;prediction accuracy
0 引言
藥物從最初的實驗研究到最終批準上市,整個階段需要13~15年,耗資20~30億美元[1]。而近兩年數據顯示,新藥研究數量與最終上市數量的比例還不到1%。因此越來越多的公司開始對現有的藥物分子進行篩選,以期挖掘出藥物的新療效,這也就是人們常說的藥物重定位。與新藥研發相比,藥物重定位只需3~5年時間,藥代動力學等不確定性顯著減小;并且,用于開展藥物重定位研究的藥物通常已經通過了臨床試驗的幾個階段或是已經上市,這使得研發成本及風險明顯降低,周期縮短。因此,藥物重定位是目前解決新藥開發高投入低成功率問題的有效方法之一[2]。
隨著藥物重定位技術的不斷發展和成熟,成功的實例也越來越多。其中具有代表性的如阿司匹林,原來用于解熱鎮痛和抗風濕,后來發現可以用于感冒、發熱、頭痛、牙痛、關節痛、風濕痛心肌梗死以及預防術后血栓的形成[3]。再如沙利度胺,原來用作鎮靜劑,后來發現能夠對惡心、失眠、孕吐產生作用。而近期研究表明,它可能適用于麻風病,多發性骨髓瘤,中到重度麻風結節性紅斑以及Ⅱ型糖尿病[4]。盡管藥物重定位蘊含著巨大潛力,但是藥物新療效的發現并不容易,因為大部分藥物的新用途可能與原本的適應癥并無明顯關系。近年來,大規模的基因組、表型數據以及藥物的化學與生物活性數據的爆炸式增長,又為藥物重定位提供了機遇。有研究人員基于藥物重定位的生物學依據,即一藥多靶和一靶多治提出了多種假設,從而實現了藥物的重定位。基于這種假設,具有相同屬性的藥物傾向于有相同或相似療效,因此可以用于治療相同或相似疾病。例如,Iwata等[5]通過比較藥物在癌細胞系上的Pathway信息,發現了可以用于抗癌的新藥物;Iorio F等[6]依據數據庫中不同的小分子藥物所影響的基因表達譜發現血管擴張藥物法舒地爾可與抗精神藥物三氟拉嗪通過組合方式治療退行性疾病;Ken等[7]通過假設作用于相似靶標或者Pathway的藥物可能會產生相似的副作用,然后通過評估8種常規阿爾茲海默癥病藥物的共同副作用,發現了25種可能可以用于該疾病的新藥物。同理,基于相似的疾病傾向于被相同或相似的藥物所治療的假設,Hu等[8]通過基因表達譜構建了疾病相似性網絡,發現躁郁癥和遺傳痙攣性截癱有著相似的發病機制,因此認為治療躁郁癥的藥物可以用于遺傳痙攣性截癱。此外,基于藥物與疾病特征之間的負相關思想,已經提出了幾種利用疾病相關特征尋找藥物新適應癥的方法。例如,Azam等[9]根據KEGG數據庫中所給出的信號通路,基于基因表達特征在疾病狀態和藥物狀態的擾動方向相反,則該藥物可以用于治療該疾病的假設,構建了全局生物分子網絡,從而發現了可能可以用于治療IPF(特發性肺纖維化)的8種新的治療藥物。還有研究人員根據藥物疾病的負相關性預測抗潰瘍藥物甲氰咪胍可以用來治療肺腺癌[10-11]。此外,基于靶標藥物重定位的基本思想即發現已有藥物的未知靶標,提出假設:如果已知藥物可以結合的靶標是其原用途以外,并且是某種疾病的關鍵分子,那么這種藥物可以用于治療該疾病。基于此假設,預測出很多藥物潛在的新靶標。例如,恩他卡朋原來是用于治療帕金森疾病的,后來發現可以作用于結核病中的結合桿菌酶,使其喪失活性,從而達到治療結核病的效果[12]。例如,致幻劑DMT與血清素受體之間的關系就是基于此種原理發現的[13]。
與之前通過單一或較少數據源提取特征相比,通過藥物的化學結構、副作用以及靶標相關數據和疾病表型數據等多種數據源提取出更多的特征,然后將藥物疾病特征根據已知的公式進行關聯整合,最后應用于新的模型,即XG-BOOST模型進行預測。
1 數據源
本文應用XG-BOOST模型預測已有藥物的潛在適應癥,具體流程如圖1所示。首先,將獲得的藥物信息與疾病信息通過計算機處理成一個個二階矩陣,矩陣中的每個元素表示藥物與藥物的相似度(或疾病與疾病的相似度),然后將每個藥物矩陣與疾病矩陣分別整合成一個個低秩矩陣,每個低秩矩陣分別表示一個特征。最后,通過這些特征數據以及參數調節獲得一個性能更優的模型,該模型可以進一步用于預測新的潛在藥物疾病關聯。
1.1 藥物疾病的黃金標準集
本次用于訓練的黃金標準集來自多個數據源,其中疾病主要來自OMIM,藥物來自DrugBank數據庫,這是一個混合的化學信息資源,具有詳細的藥物數據和全面的目標信息。通過使用UMLS(Unified Medical Languange System)將藥物、藥物的適應癥和疾病名稱進行匹配。為確保關聯的可信度,要求藥物與疾病必須有多種關聯,最終得到包括1 244種藥物疾病關聯的黃金標準集,其中包括來自DrugBank數據庫的443種藥物,以及OMIM中所列舉的256種疾病。
1.2 藥物疾病相似性
1.2.1 藥物相似性
定義四組衡量藥物與藥物相似策略:
(1)化學相似性。藥物的SMILE分子式可以直接從DrugBank數據庫中獲得,通過RDKit數據包處理成分子指紋,最終獲得的藥物和藥物相似性得分是基于指紋的二維Tanimoto系數。
(2)副作用相似性。藥物的副作用通過SIDER庫獲得,這是個線上開源的數據庫,每個藥物副作用之間相互獨立,沒有關聯,因此可以通過Jaccard系數求得藥物之間的相似性,即用交集比上并集。
(3)ATC相似性[14]。ATC代碼共有7位,其中第1、4、5位是字母,第2、3、6、7位為數字。ATC系統將藥物分為5個級別,分別為解剖學、治療學、藥理學、化學、化合物上的分類。兩種藥物成分的K級藥物療效相似性定義為:
(4)Go相似性。藥物相關的基因語義相似性得分通過軟件GoSemSim獲得,基因之間的功能相似性廣泛應用于生物信息學,Go相似性則主要用于評估基因之間的功能相似性。
(5)ppi相似性。藥物靶蛋白相互作用的相似性主要是通過已知靶蛋白之間的相互作用構建出人類靶蛋白相互作用網絡,其中靶蛋白相互作用關系可以從HPRD數據庫中獲得,該數據庫也是一個在線的開源數據庫。通過計算出兩個靶蛋白之間的最小距離,將最小距離進行歸一化從而得到這兩個靶蛋白的相互作用得分。將兩個藥物對應的靶蛋白兩兩作用后求出得分平均值,即為兩個藥物的ppi相似性。
1.2.2 疾病相似性
對于疾病,主要采用疾病的表型特征,使用的疾病相似性由van Driel等[15]構建。疾病相似性由基于疾病表型的MinMiner計算獲得,其計算類似于廣泛用于信息檢索的術語頻率—逆文檔頻率技術。簡而言之,通過使用醫學主題標題詞匯(Mesh)的解剖學(A)和疾病(C)部分,將每種疾病描述為特征向量,以從其OMIM記錄中自動提取Mesh術語,其中特征向量中的每個值表示Mesh概念與表型的相關性。每個概念的相關性是通過文檔中概念的實際計數加上概念下位詞的相關性總和計算得到。一對疾病{di,dj}之間的相似性通過計算兩個網格概念向量之間的余弦相似性ti={ti1,ti2,…,tik}和tj={tj1,tj2,…,tjk}得到。為了計算藥物化學相似性,將每種化合物描述為基于PubChem指紋880維的二維向量,其中如果相應的指紋包含在藥物中,則向量中的元素為1,否則為0。用Tanimoto系數計算出兩種化合物之間的二維相似度,其定義為普通指紋數與指紋總數的比率。
1.2.3 藥物疾病整合
將藥物的4種相似性與疾病mesh相似性整合成5個特征,即特征1為藥物化學結構相似性-mesh相似性,特征2為藥物副作用相似性-mesh相似性,特征3為藥物atc相似性-mesh相似性,特征4為Go相似性-mesh相似性,特征5為藥物ppi相似性-mesh相似性。對于一對藥物疾病的關聯得分(dr,di),計算步驟如下:首先,將之前得到的黃金標準集中的每一對關聯表示為(dr,di),計算所求藥物和已知藥物之間的相似性S(dr,dr)與所求疾病和已知藥物關聯的疾病之間的相似性S(di,di)。根據Perlman等[16]提出的方法,將這兩個相似性得分通過計算加權平方根合并成一個相似性得分。
以特征1為例,需將某種藥物m與黃金標準集中的藥物n求基于化學結構的相似性得分,然后求出該藥物m對應的疾病與藥物n對應疾病基于mesh的相似性得分,這樣會得到1 244個相似性數據。選取最大的一個作為該組藥物疾病的關聯得分,并以相同的方法求出另外幾組特征。
1.3 數據集
數據集的正集由黃金標準集組成,為了完善特征數據,事先刪除存在數據缺乏的藥物。最終標準集包括1 244組藥物疾病對,負集的數據量是正集的兩倍大小,它由隨機產生的藥物疾病對構成。簡而言之,就是在所有185 609個藥物疾病對中,去掉正集,剩下隨機抽取3 866個作為負集。
2 XG-BOOST模型
2.1 監督學習
監督學習就是訓練帶標簽數據的學習。比如,有10萬條數據,每條數據都包括50個特征,還有一個標簽。而標簽的內容取決于學習問題,如果數據是病人進行癌癥診斷做的各項檢查結果,標簽就是病人是否得癌癥,是為1,否為0。監督學習就是要從這10萬條數據中學習根據檢查結果診斷病人是否得癌癥的知識,而學習的范圍就限定在這10萬條數據中,形象的理解就是,在這10萬條帶標簽數據的“監督”下進行學習。
2.2 模型原理
XG-BOOST是一個監督學習模型,它是多個CART樹(分類回歸樹)組合后的模型,這種組合后的模型一般都具有更強大的泛化能力[17-19]。因此,XG-BOOST模型最終的預測值就是每棵樹的預測值之和。對于分類問題,由于CART樹的葉子節點對應的值是一個實際分數,而非一個確定的類別,這將有利于實現高效的優化算法,這也是XG-BOOST運算快的原因所在。該模型的數學表示如下:
K是指樹的棵數,F表示所有可能的CART樹,f表示一個具體的CART樹,整個模型由K個CART樹組成。
模型表示出來后可進一步表示出模型的目標函數,如式(4)所示。
該目標函數包含兩部分,第一部分是損失函數,第二部分是正則項,正則項由K棵樹的正則化項相加而來。
2.2.1 目標函數
訓練該模型的任務就是最小化目標函數,尋找一組最佳參數組。XG-BOOST模型的參數優化不是直接優化整個目標函數,而是分步驟優化目標函數,先優化第一棵樹,再優化第二棵樹,直至最后一顆樹。過程如下:
2.2.2 正則化項
對于一棵樹的正則化,作出如下定義:
首先,一棵樹有T個葉子節點,這T個葉子節點的值組成了一個T維向量w,q(x)是一個映射,用來將樣本映射成1~T的某個值,也即它分到某個葉子節點,q(x)其實代表了CART樹的結構,w_q(x)自然就是這棵樹對樣本x的預測值。
有了上述定義,XG-BOOST的正則化項如下:
這里的參數[γ]和[λ]都是XG-BOOST自己定義的,在使用該模型時,這兩個參數可以自己調節,[γ]越大表示希望獲得結構越簡單的樹,因此對較多葉子節點的樹懲罰越大。[λ]越大也表示希望獲得結構越簡單的樹。
3 結果分析
運用上述方法預測潛在的藥物疾病關聯,并將該方法與邏輯回歸、隨機森林進行比較。
3.1 性能評估
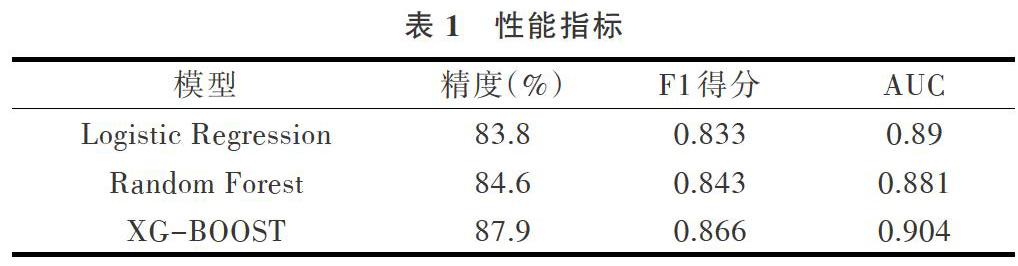
K折交叉驗證常用的是5、10、20折,在折數選擇問題上,不同折數的計算成本和效果均存在差異,因此需要進行權衡。本文采用5折交叉驗證,并通過AUC、F-measure及精度等常用性能指標對模型進行評價,具體性能指標如表1。
3.2 性能分析
進一步繪制得到AUC,即ROC曲線面積,其中橫軸表示真負率,縱軸表示真正率或靈敏度。在整個預測過程中,正樣本被預測出來的比率越大越好,與之對應的負樣本被預測為正樣本的比率越小越好,也即ROC曲線越靠近坐標系左上角,分類效果越好。根據圖2可以看出,XG-BOOST的性能最好,隨機森林次之,最后是邏輯回歸[20]。XG-BOOST部分預測結果如表2所示。
4 結語
傳統新藥開發耗時長且風險高,舊藥新用作為另一種研究范式受到廣泛關注。近年來,隨著藥物和疾病數據的快速積累,挖掘各層次生物醫學數據預測藥物新用途成為系統生物領域的研究熱點。本文從藥物的化學結構、藥理性質、副作用、靶蛋白功能和疾病表型等數據中挖掘出有信息量的特征,然后應用于XG-BOOST模型,并與邏輯回歸、隨機森林模型性能作對比。實驗結果表明,本文方法性能更優,原因在于其整合利用了藥物化學結構、靶蛋白、藥理、表型等各層次數據;在特征構建完后,采用XG-boost模型。
但該方法仍有一些問題待研究解決,如數據的黃金標準集是通過使用UMLS將Drugbank中的藥物、藥物適應癥和OMIM中的疾病名稱進行匹配得到的,因此獲得的藥物疾病關聯不完善;并且負集是隨機產生的一個兩倍大的集合,這會存在許多不確定性。隨著更多關聯的產生,預計可以構建更多具有生物學意義的集合,使得預測結果更準確。
參考文獻:
[1] 楊光,郝逸凡. 基于互信息法的抗前列腺癌藥物重定位分析[J]. 沈陽師范大學學報:自然科學版,2019(1):34-37.
[2] 劉艷飛,孫明月. 網絡藥理學在中藥研究中的應用現狀與思考[J]. 中國循證醫學雜志,2017(11):1344-1349.
[3] 張永祥. 藥物重定位——網絡藥理學的重要領域[J]. 中國藥理學與毒理學雜志,2012(6):779-786.
[4] WANG Y Y,CUI C F,QI L Q,et al. DrPOCS:Drug repositioning based on projection onto convex sets[J]. IEEE/ACM Transactions on Computational Biology & Bioinformatics,2019,16(1): 154-162.
[5] IWATA M,HIROSE L. Pathway-based drug repositioning for cancers: computational prediction and experimental validation[J].? Medical Chemtsry,2018, 61:9583-9595.
[6] IORIO F. Discovery of drug mode of action and drug repositioning from transcriptional responses[J].? Proc Natl Acad Sci. USA,2010,107(33): 14621-14627.
[7] MCGARRY K, GRAHAM? Y. RESKO: Repositioning drugs by using side effects and knowledge from ontologies[J].? Knowledge-based Systems,2018,160: 34-48.
[8] HU G,AGARWAL P. Human disease-drug network based on genomic expression profiles[J].? PLoS One,2009,4(8): 6536.
[9] NAFISEH A. A novel computational approach for drug repurposing using systems biology[J]. Bioinformatics,2018,34(16):2817-2825.
[10] LI Y. Gene expression module-based chemical function similarity search[J]. Nucleic Acids Res,2008,36(20): 137.
[11] WANG G. Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma[J]. PLoS One,2011,6(1): 14573.
[12] KINNINGS S L. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis[J]. PLoS Comput Biol.,2009,5(7): 1000423.
[13] KEISER M J. Relating protein pharmacology by ligand chemistry[J]. Nat Biotechnol,2007,25(2): 197-206.
[14] 陳范曙. 基于信息整合的藥物相關信息挖掘方法研究[D]. 2016.
[15] VANDRIEL M A. A text-mining analysis of the human pheome[J]. Eur J Hum Genet,2006,14:535-542.
[16] ASSAF G, GIDEON Y. PREDICT: a method for inferring novel drug indications with application to personalized medicine[J]. Molecular System Biology, 2011,7(496).
[17] MASON L, BAXTER J,BARTLETT P L.Boostig algorithms as gradient deacent[C]. Conference on Advantage in Neural Information Processing Systems,2000:512-518.
[18] DIOGO M,KATHERINE M. Next-generaion machine learning for biological network[J]. Cell,2018,173:1581-1592.
[19] KHADER S, K W J, B S G,et al. priortizing small molecule as candinates for drug repositioning using machine learning[EB/OL].? https://www.biorxiv.org/content/10.1101/331975v1,2018.
[20] 王博. 基于Logistic Regression的數學成績預測系統的研究[D]. 南昌:南昌大學,2018.
(責任編輯:孫 娟)
