小動脈木村病1例
2020-06-29 09:15:46宣瑞紅劉小麗
臨床與實驗病理學雜志 2020年5期
宣瑞紅,劉小麗
患者男性,76歲,左眉弓上方皮下小動脈外膜腫物伴瘙癢1年余,為明確診斷,行手術活檢。體檢未見明顯異常。術中見左眉弓上方皮下腫物呈梭形,手術完整切除。
病理檢查眼觀:送檢皮下組織1塊,大小2.2 cm×0.5 cm×0.3 cm,腫物界限清楚,未見明顯包膜,切面灰紅色,實性質中。其內可見2處直徑小于0.1 cm的管腔樣結構,與周圍組織黏連。鏡檢:多處直徑0.05~0.1 cm的小動脈周圍淋巴組織增生(圖1),淋巴濾泡形成(圖2),破壞小動脈外膜;毛細血管內皮細胞增生,形成肉芽腫樣結構(圖3);嗜酸性粒細胞浸潤形成嗜酸性微膿腫(圖4),并伴不同程度的纖維化(圖5)。免疫表型:生發中心及少量套區細胞CD20陽性(圖6),生發中心濾泡樹突細胞CD21陽性,生發中心B細胞CD79a弱陽性,濾泡其余細胞陽性,濾泡生發中心Ki-67高表達,其余區域Ki-67增殖指數均較低,均顯示濾泡結構存在。
病理診斷:嗜酸性粒細胞增生性淋巴肉芽腫。
討論木村病是一種特殊的淋巴組織增生性疾病,也稱為嗜酸性粒細胞增生性淋巴肉芽腫,最早由中國學者金顯宅提出,1948年日本學者Kimura再次報道。該病好發于亞洲人,通常累及頭頸部皮下組織、淋巴結、口腔、腋窩、腹股溝、四肢和軀干等部位,眼眶、眼瞼和眼外肌發病文獻報道較少[1]。該病病程較長,中青年男性占主導地位,男女比為3.5 ∶1,可單發也可多發,常伴瘙癢及外周血嗜酸性粒細胞增高及免疫球蛋白IgE水平升高[2],無其他不良反應,病因尚不明確,可能的因素包括過敏、感染、節肢動物咬傷、T細胞調控異常、嗜酸性粒細胞動力學異常和IgE合成異常。手術切除常有復發,非甾體消炎藥、類固醇激素、冷凍療法、化療、放療、激光治療可作為術后輔助治療[3]。
病理學檢查是診斷該病的關鍵,表現為大量淋巴細胞浸潤,形成淋巴濾泡,大量的嗜酸性粒細胞浸潤,形成嗜酸性微膿腫,血管內皮細胞增生,形成肉芽腫樣結構,不同程度的纖維膠原組織增多[4]。
鑒別診斷:木村病需與其他淋巴組織增生性病變鑒別,主要是血管淋巴樣增生伴嗜酸性粒細胞增多癥(angiolym-phoid hyperplasia with eosinophilia, ALHE),又稱上皮樣血管瘤,該病更少見,常見部位是頭頸部,屬于良性血管腫瘤[5],主要組織學特征是大小不等的血管增生,且內皮細胞胞質豐富、腫脹,形成上皮樣或組織細胞樣結構,周圍浸潤的炎細胞以T淋巴細胞為主,淋巴濾泡形成較少,其本質為良性血管瘤,免疫組化標記CD3、CD34均陽性,CD20陰性。木村病毛細血管內皮細胞增生,形成肉芽腫結構,增生的是薄壁血管,且非腫瘤性增生;炎癥細胞主要是B淋巴細胞為主,免疫組化標記CD20、CD79a陽性,伴有嗜酸性粒細胞,且可形成淋巴濾泡,免疫組化標記能清楚顯示濾泡結構。
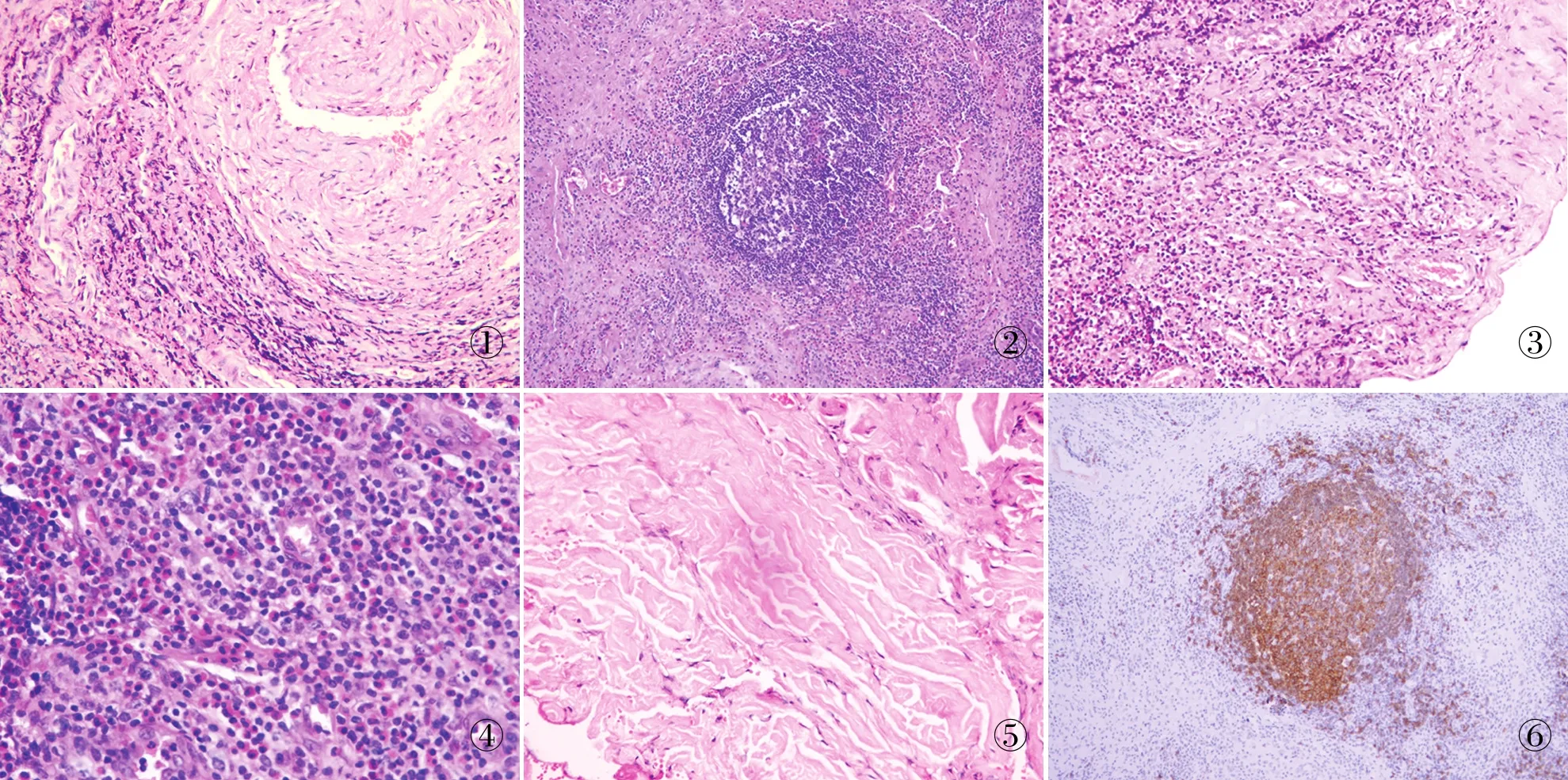
圖1 小動脈周圍可見淋巴細胞廣泛浸潤 圖2 淋巴濾泡形成 圖3 血管內皮細胞增生,形成肉芽腫樣結構 圖4 大量的嗜酸性粒細胞浸潤,形成嗜酸性微膿腫 圖5 伴不同程度的纖維化 圖6 生發中心及少量套區細胞CD20陽性,顯示濾泡結構存在,EnVision法
另外,有文獻報道木村病可導致腎病綜合征,誘發可逆性的繼發性膜性腎病,單純激素治療效果不明顯,隨著腫塊增大,蛋白尿和氮質血癥均加重。手術切除腫塊后,腎臟功能可恢復正常。該表現可能由未知的分泌分子引起[6-7]。系膜增生、嗜酸性細胞浸潤和足突融合是這類患者的顯著組織學特征。患者對強的松治療敏感,但易復發。局部切除術有助于防止復發[6]。
有學者對木村病的細胞學進行分析,其鏡下表現與組織學檢查結果相關。細胞學特征包括:大量淋巴細胞、大量嗜酸性粒細胞、膠原組織碎片、內皮細胞、偶見的多核細胞。但此類細胞學特征并非木村病特有,其他腫瘤可能有類似的疾病表現及細胞學表現。在診斷木村病之前必須排除這些因素。探討木村病的細胞學特點需緊密結合臨床病理學特征[8]。
