廢晶硅太陽能電池資源化分類回收技術研究*
2020-07-01 02:27:44馮晉堯劉景洋周瀟云畢瑩瑩曲豐作
環境污染與防治 2020年6期
董 莉 馮晉堯, 劉景洋# 周瀟云 畢瑩瑩 曲豐作
(1.中國環境科學研究院,國家環境保護生態工業重點實驗室,北京 100012; 2.大連工業大學輕工與化學工程學院,遼寧 大連 116034)
各種太陽能電池中,晶硅太陽能電池技術目前發展最成熟,在應用中占主導地位[1]。2012年1月,歐盟將光伏組件列入廢棄電氣和電子設備(WEEE)規章,廢棄太陽能光伏組件必須進行收集和回收利用。據中國光伏協會統計,在2018年,中國光伏組件新增裝機容量43 GW,累計裝機容量超過170 GW,新增和累計裝機容量均為全球第一。據中國可再生能源協會預測,到2030年,中國廢棄的光伏組件將包含145萬t碳鋼、110萬t玻璃、54萬t塑料、26萬t鋁、17萬t銅、5萬t硅和550 t銀。
國內外學者對廢電池片中鋁、銀、硅等資源回收進行了相關研究。使用硝酸和過氧化氫混酸,在一定溫度下,能將電池表面的鋁和銀涂層全部浸入溶液體系中,但兩種金屬存在于同一溶液對金屬后續的分離回收造成一定的困難[2-8]。在不同溫度下,可采用不同濃度的氫氧化鉀溶液回收鋁,并采用3種以上不同濃度的混酸蝕刻防反射層和P-N結,但氫氧化鉀會對晶硅進行蝕刻,造成一定量硅損失,且3種以上混酸的體系較為復雜,加大了對酸液的回收處理難度[9-11]。據此,本研究在室溫下對廢晶硅太陽能電池中銀、鋁和硅進行分類、分步資源化回收,建立一套硅質量損失較少、溶液體系簡單、有利于溶液中的元素資源化利用的回收技術。
1 實驗部分
1.1 實驗材料
典型晶硅太陽能電池片結構見圖1。從上到下分別為正面電極、防反射層、N型層、硅基片和背面電極。正面電極一般為銀柵線電極(覆蓋導電銀漿),防反射層主要為氮化硅,N型層是在硅基片內摻雜磷元素,與硅基片形成P-N結傳輸電子,背部電極一般為刷滿整面的鋁漿料(即鋁電極)[12]。實驗所用廢晶硅太陽能電池尺寸為15 cm×15 cm,厚度為(180±15) μm,單片晶硅太陽能電池(以下簡稱電池片)的質量在10.5 g左右。
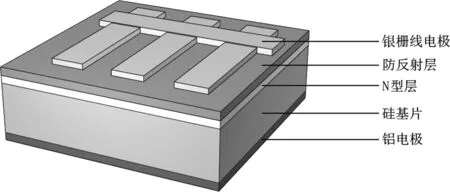
圖1 晶硅太陽能電池結構Fig.1 Crystalline silicon solar cell structure
1.2 實驗方法
將電池片先用去離子水清洗,烘干。取4份完整電池片稱重,在室溫下分別浸入體積分數為10%、20%、30%、40%的鹽酸中反應,在不同時間段取樣,通過電感耦合等離子體原子發射光譜法(ICP-AES)測定溶液中鋁濃度。
將去除鋁涂層的電池片在室溫下分別浸入體積分數為20%、25%、30%、35%、40%的硝酸中反應,在不同時間段取樣,通過原子吸收光譜法(AAS)檢測溶液中銀濃度。
配制體積分數分別為40%-4%、35%-6%、40%-6%、45%-6%和40%-8%的硝酸-氫氟酸混酸,將去除鋁和銀涂層的電池片在室溫下分別浸入上述配比的混酸中進行反應,反應后所殘留的固體為單質硅。通過硅元素滴定法[13]分析不同時間內溶液中硅濃度。
對反應后的電池片進行清洗,烘干,選擇典型時間節點的電池片分別采用掃描電子顯微鏡(SEM)、能譜(EDS)方法進行分析。
鋁、銀浸出率及硅回收率的計算公式如下:

(1)

(2)
2 結果與討論
2.1 鋁浸出結果
圖2為鋁浸出率曲線。反應初始階段,鋁涂層溶解速率較快,一定時間后鋁涂層溶解速率急劇減緩,最終停止。縱觀4個濃度下的反應情況,鹽酸體積分數越高,鋁完全浸出時間越短(見表1)。鹽酸體積分數大于等于20%,反應速率差異不大;小于10%,反應速率較為緩慢。故選擇體積分數為20%的鹽酸反應時間40 min作為鋁浸出的最佳條件,浸出率可達99.77%。此外,所得的氯化鋁浸提液可用于水處理中的絮凝劑或者進一步加工生產聚合氯化鋁[14-17]。
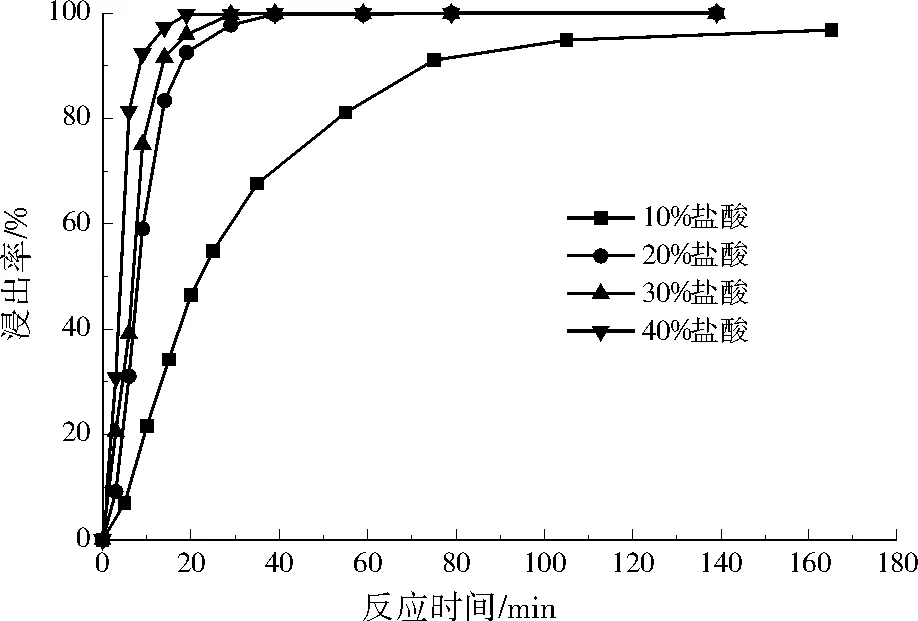
圖2 不同體積分數鹽酸的鋁浸出率Fig.2 Aluminum leaching rate in hydrochloric acid solution of different volume fractions

表1 鋁浸出結果
對20%鹽酸浸出鋁的過程進行對比分析。反應開始前,電池片背面布滿了鋁涂層(見圖3(a));反應20 min時,有少量鋁涂層殘留在電池片背面,鋁涂層與硅基片的交接邊緣顯現(見圖3(b));反應40 min時,電池片無鋁涂層覆蓋,鋁已全部浸出。對20%鹽酸反應40 min后的電池片背面進行EDS分析,證實鋁涂層已無殘留。
2.2 銀浸出結果
圖4為銀浸出率曲線。反應初始階段,銀浸出速率較快,一定時間后速率急劇減緩,最終停止反應。硝酸體積分數越高,銀完全浸出時間越短(見表2)。體積分數為20%的硝酸中,銀浸出速率很緩慢,150 min時仍未檢出反應終點。當硝酸體積分數為25%和30%時,銀完全浸出時間分別為90、70 min,浸出時間依然較長。硝酸體積分數從35%提高到40%,雖然也能加速銀的浸出,但增加效果有限,這與金懷東等[18]的研究結果一致。硝酸濃度太高,易揮發;硝酸濃度太低,不利于銀的溶解。綜合實驗結果,選擇體積分數為35%硝酸反應30 min作為銀浸出最佳反應條件,銀最終浸出率為99.60%。所得的以硝酸銀為主的浸提液可用于生產硝酸銀或回收提取單質銀[19-22]。

圖3 電池片在20%鹽酸中反應前后的SEM圖(×15 000)Fig.3 SEM images of cells before and after leaching in 20% hydrochloric acid (×15 000)
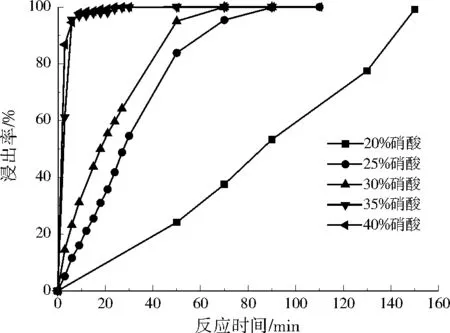
圖4 不同體積分數硝酸的銀浸出率Fig.4 Silver leaching rate in nitric acid solution of different volume fractions

表2 銀浸出結果
反應開始前,電池片正面電極導電銀漿突起且明亮(見圖5(a));在反應過程中,銀漿逐漸被蝕刻,形成凹陷,顏色暗淡,出現凹槽;反應30 min時,電池片正面電極僅剩凹槽痕跡,無銀漿光澤存在(見圖5(b))。對35%硝酸反應30 min后的電池片進行EDS分析,發現無銀殘留。

圖5 電池片在35%硝酸中反應前后的SEM圖(×15 000)Fig.5 SEM images of cells before and after leaching in 35% nitric acid (×15 000)
2.3 硅回收結果
由圖6可見,當混酸中氫氟酸體積分數為4%時,反應時間較長;而混酸中氫氟酸體積分數為8%時,反應過程中有冒黃煙現象,且高濃度氫氟酸腐蝕性較強,對晶硅表面會造成損傷,單質硅損失較大。故混酸中氫氟酸體積分數選擇6%。

圖6 混酸去除防反射層和N型層時的硅損失率變化Fig.6 Silicon loss rate in mixed acid removal of anti-reflection layer and N-type layer
在混酸中氫氟酸體積分數確定為6%的前提下,硝酸體積分數為35%時,防反射層硅脫除速率較慢,到120 min左右才能夠將N型層蝕刻完成;硝酸體積分數為45%時,防反射層硅脫除速率較快,但過早進入N型層反應階段,致使硅損失率在30%以上。綜合硅損失和反應時間,故混酸配比選擇40%硝酸-6%氫氟酸。
結合圖7,回收單質硅經歷了3個重要階段。第1階段(反應0~35 min),氮化硅完全浸出,防反射層去除;第2階段(35~75 min)為N型層的脫除;第3階段(75~140 min)為晶硅的過度蝕刻,此時單質硅開始損失。通過EDS分析發現,反應75 min后,電池片的防反射層及N型層已經蝕刻干凈,僅剩單質硅。因此,選擇40%硝酸-6%氫氟酸作為浸出液,反應75 min能完成對防反射層及N型層的去除,硅回收率為85.51%。單質硅可作為多晶硅錠的生產原料。

圖7 電池片在40%硝酸-6%氫氟酸中反應前后的SEM圖(×15 000)Fig.7 SEM images of cells before and after leaching in 40% nitric acid-6% hydrofluoric acid (×15 000)
2.4 資源化回收路線
結合2.1節至2.3節,得到了氯化鋁浸提液、硝酸銀浸提液和單質硅,實現了對廢晶硅太陽能電池的分類、分步資源化回收,得到完整的技術路線,見圖8。
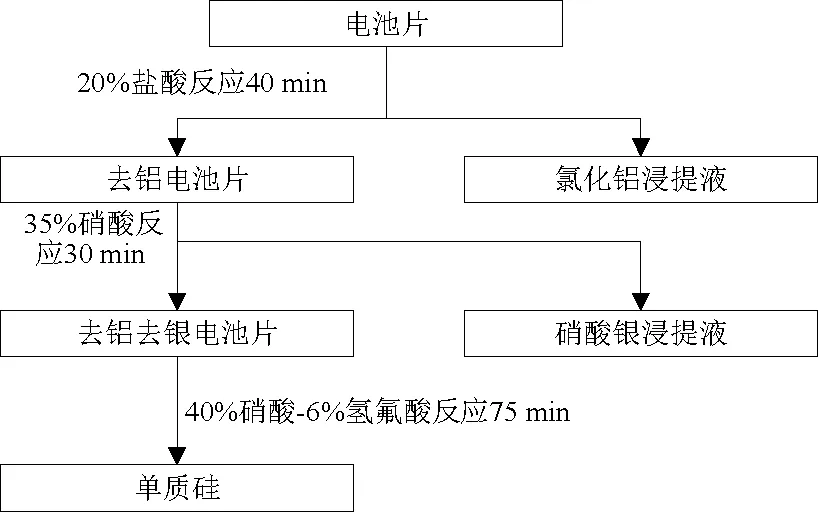
圖8 電池片資源化分類回收技術路線Fig.8 Classification and recycling technology roadmap of cell
3 結 論
(1) 鋁的浸出反應速率隨著鹽酸濃度升高而加快。鋁的資源化回收最佳參數為20%鹽酸反應時間40 min,鋁浸出率為99.77%。
(2) 隨著硝酸濃度升高,銀的浸出速率加快。銀的資源化回收最佳參數為35%硝酸反應時間30 min,銀浸出率為99.60%。
(3) 回收單質硅經歷防反射層氮化硅的去除、N型層的去除及晶硅的過度蝕刻3個階段,硅資源化回收最佳參數為40%硝酸-6%氫氟酸反應時間75 min,硅回收率為85.51%。
(4) 廢晶硅太陽能電池中鋁、銀及單質硅進行資源化分類回收后,氯化鋁可作為水處理的絮凝劑或生產聚合氯化鋁,硝酸銀可進一步提取單質銀或生產硝酸銀,單質硅可作為多晶硅錠的生產原料。
