反相高效液相色譜法測定神經酸片劑中神經酸含量
2020-07-04 01:42:58吳樂艷侯雯清孫孔春楊璨瑜沈報春
昆明醫科大學學報 2020年6期
吳樂艷,侯雯清,孫孔春,楊璨瑜,舒 紋,沈報春
(昆明醫科大學藥學院暨云南省天然藥物藥理重點實驗室,云南昆明 650500)
神經酸(Nervonic acid) 因最早發現于哺乳動物的神經組織中而得名,其在周圍神經組織和腦組織中含量較高,是神經組織中生物膜的重要組成成分,也是大腦發育及維持正常功能所必須的營養物質,對神經介質和受體發揮作用具有重要的意義[1]。有研究表明,神經酸具有增強體液免疫和細胞免疫[2]、改善記憶、認知功能損傷保護和抗氧化[3]作用,能疏通受損大腦神經通路并可促進神經細胞再生[4]。此外,神經酸對帕金森病[5-6]、腦白質疏松癥[7-8]、Zellweger 綜合征[9]、多發性硬化癥[10]、腎上腺腦白質營養不良[11]、腦血管疾病[12-13]等有一定的治療和預防作用。神經酸被認為是21 世紀最有前途的腦健康保健品和藥品,為預防和治療腦部疾病提供了一條新途徑,具有廣闊的應用前景[14]。
目前,神經酸定量分析多采用衍生化氣相色譜法[15-18],該方法是將神經酸酯化后測定酯化物的含量,前處理過程較繁瑣,且可能存在未反應的神經酸,導致神經酸含量測定不準確。文獻[19]采用了高效液相色譜法測定蒜頭果油分離出的神經酸含量,本文將建立一種反相高效液相色譜方法直接測定神經酸的含量,該方法不需對樣品進行酯化,可直接進樣分析,流動相組成簡單,分析時間短,定量準確,可在短時間內實現樣品的快速分析。
1 材料與方法
1.1 儀器與試劑
日本島津LC-2010AHT 高效液相色譜儀(包含島津SPD-M10A 檢測器,島津LC solution 色譜工作站);超純水機(Direct-Q 3 UV):美國Millipore 公司;電子天平(BS224S):北京賽多利斯儀器系統有限公司;超聲清洗儀(SK3200H):上海科導超聲儀器有限公司;神經酸標準品:由云南盈木佳生物科技有限公司提供(純度99.0%);神經酸片劑:某公司生產;乙腈(色譜純):上海星可高純溶劑有限公司;磷酸(分析純)。實驗用水為超純水(電阻率為18.2 MΩ·cm,25℃)。
1.2 色譜分析條件
1.3 標準溶液配制
精密稱取神經酸標準品0.050 4 g,以乙腈為溶劑,配制濃度約為2 mg/mL 的標準儲備液。精密量取神經酸標準儲備液1 mL 至10 mL 容量瓶中,加入9 mL 乙腈定容至刻度,搖勻,即得濃度為0.2 mg/mL 的標準溶液;再按照濃度梯度稀釋法分別配制濃度為0.1 mg/mL、0.05 mg/mL、0.02 mg/mL、0.01 mg/mL 的多份標準溶液。
1.4 待測樣品溶液配制
取神經酸片劑10 片,精密稱定,研細。精密稱取適量,置10 mL 容量瓶中,加乙腈適量,超聲處理使主成分溶解,乙腈定容至刻度,充分搖勻,得待測樣品溶液,備用。
1.5 測定
將標準溶液及待測樣品溶液先用0.22 μm 的有機系濾膜過濾,取續濾液以10 μL 的進樣量上樣于高效液相色譜儀,記錄保留時間、峰面積等參數。按外標標準曲線法以峰面積計算,即可測得神經酸片劑中神經酸的含量。
2 結果
2.1 方法學驗證
2.1.1 標準曲線精密吸取神經酸系列標準溶液各10 μL,照“1.2.1”項下色譜條件進樣分析,以各份標準品溶液的濃度(X,mg/mL) 為橫坐標,以神經酸色譜峰的峰面積(Y) 為縱坐標作圖(見圖1),得線性回歸方程y=3 000 000 X(mg/mL)+4 795.9,R2=0.999 9。神經酸含量在0 .01~0.2 mg/mL 濃度范圍內具有良好的線性關系(P=0.000)。
2.1.2 精密度實驗精密吸取2 mg/mL 神經酸標準儲備液10 μL,照“1.2.1”項下色譜條件,連續進樣6 次,記錄峰面積,結果見表1。峰面積相對標準偏差RSD 為1.80%,說明實驗方法精密度良好。
2.1.3 穩定性實驗精密吸取待測樣品溶液10 μL,照“1.2.1”項下色譜條件,在24 h 內測定6次(0 h、2 h、4 h、8 h、12 h、24 h),記錄峰面積,結果見表1。由表1可見,峰面積相對標準偏差RSD為1.52%,待測樣品溶液在24 h內穩定性良好。
2.1.4 加樣回收率照“1.2.1”項下色譜條件,首先測定待測樣品溶液中神經酸的峰面積,帶入“2.1.1”項下標準曲線,求得待測樣品溶液的濃度。接著將待測樣品溶液平均分成9 份,再分成3 組,每組分別添加相當于待測樣品溶液中神經酸含量80%、100%、120%三個水平的神經酸標準溶液,混勻后照“1.2.1”項下色譜條件分析,結果見表2。由表2 可見,添加神經酸標準溶液后所得樣品的回收率在96.94%~104.46%之間,峰面積相對標準偏差RSD 在0.81%~3.80%之間,平均回收率為101.53%。該方法準確度良好,可用于神經酸片劑中神經酸的定量分析。
2.2 樣品含量測定

圖1 神經酸線性回歸方程Fig.1 Linear regression equation of nervonic acid

表1 精密度實驗、穩定性實驗結果(1)Tab.1 Precision and stability results (1)

表1 精密度實驗、穩定性實驗結果(2)Tab.1 Precision and stability results (2)

表2 加樣回收率實驗結果Tab.2 Spiked recoveries and RSDs results

圖2 標準品和樣品典型色譜圖Fig.2 Representative chromatograms of standard and sample
按照“1.2.3”項下方法,分別精密稱取約相當于神經酸10 mg 的片粉,制備兩份神經酸樣品溶液,采用建立的高效液相色譜方法測定兩份樣品溶液中神經酸的含量,典型色譜圖見圖2,測定結果見表3。
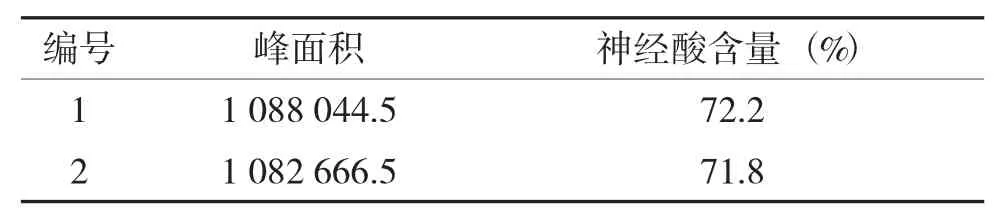
表3 樣品測定結果Tab.3 Analytical results for the sample
3 討論
神經酸不僅可以修復受損的神經纖維,而且還可促進受損的神經細胞再生,對動脈粥樣硬化、高血脂、腦梗塞、腦出血、腦萎縮、癡呆、小兒腦癱、癲癇、帕金森病、腦外傷等疾病的防治有一定作用。國外對神經酸的研究已開展了將近一個世紀,但在我國尚處于起步階段,對于其來源、獲取方法、藥理作用、生理作用及新劑型、應用等的研究還有很大空間。
本課題組在前期實驗中曾采用現有報道的衍生化氣相色譜法測定神經酸片劑中神經酸含量,先將樣品進行脂肪酸甲酯化,再用有機溶劑進行提取,氣相色譜法測定,外標法定量。實驗過程中發現由于神經酸沸點較高,采用將神經酸進行甲酯化后再定量的方法,可能存在神經酸甲酯化不完全的問題;甲酯化后又經過正己烷提取的操作,整個含量測定過程前處理操作步驟較多,會存在神經酸損失導致測定值比真實值偏小、定量不準確的問題,提取回收率較低;前處理過程復雜也存在方法的重復性及可操作性不強等缺陷。基于此,筆者建立了一種直接測定樣品中神經酸含量的反相高效液相色譜方法,提高了神經酸定量的準確度。
本研究建立的含量測定方法,首次采用C8柱,神經酸出峰時間大概在20 min,具有分析時間短,分析效率高等優點[20],實現了對神經酸片劑中神經酸含量的準確測定,該方法不需要進行衍生化,前處理過程簡單易操作,分析條件溫和,分析速度快,定量結果準確。此外,該方法的色譜條件、流動相組成簡單,精密度、重復性及RSD符合相關規定,穩定性好,靈敏度高,選擇性好。可實現人力、物力、財力的節省,有利于神經酸資源的開發及運用,具有較好的應用前景。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
兒童故事畫報(2019年5期)2019-05-26 14:26:14
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
