輸油站場(chǎng)工藝管道腐蝕失效行為分析研究
2020-07-11 07:13:04溫素麗劉翔李紅波張亞明馬瑩梁昌晶
油氣田地面工程 2020年7期
關(guān)鍵詞:分析
溫素麗 劉翔 李紅波 張亞明 馬瑩 梁昌晶
1中國(guó)石油華北油田公司第五采油廠
2中國(guó)石油華北油田公司第三采油廠
3中國(guó)石油華北油田公司二連分公司
4中國(guó)石油渤海鉆探第二鉆井工程分公司
近年來,隨著我國(guó)油田的深入開發(fā)和經(jīng)濟(jì)的快速發(fā)展,各類油氣管道的總里程不斷增加,但管道的服役環(huán)境也在不斷變化,很多管道的服役年限已超過10 年,腐蝕狀況嚴(yán)重。據(jù)統(tǒng)計(jì)數(shù)據(jù)表明,由腐蝕引起的管道失效占總失效次數(shù)的40%[1-2],在諸多腐蝕類型中,因內(nèi)腐蝕導(dǎo)致的管道失效時(shí)有發(fā)生,已成為危害管道安全、影響管道完整性的重要因素之一。南京輸油站所轄的長(zhǎng)興、嵐山和儀征站場(chǎng)內(nèi)的輸油管道發(fā)生了多次腐蝕穿孔事故[3],金陵油庫(kù)發(fā)生過多起內(nèi)腐蝕穿孔事故[4],塔河油田某站外輸油管道底部發(fā)生過多起泄漏事故[5]。這些事故都發(fā)生在輸油管道的靜置段,即液體長(zhǎng)期不流動(dòng)的管段,腐蝕形態(tài)多為局部小孔腐蝕。
某輸油站場(chǎng)管道輸送介質(zhì)為油、氣、水三相,管徑219.1 mm,壁厚8.5 mm,管材為X65 管線鋼,設(shè)計(jì)壓力1.6 MPa,實(shí)際運(yùn)行壓力0.5~0.8 MPa,運(yùn)行溫度45 ℃,外防腐采用熔接環(huán)氧粉外加犧牲陽(yáng)極陰極保護(hù)。在清理開挖土方時(shí)發(fā)現(xiàn)管道下部出現(xiàn)腐蝕穿孔,穿孔段為管道靜置段(原連接過濾器設(shè)備,現(xiàn)已拆除,三通后形成流動(dòng)死區(qū))。針對(duì)這一典型穿孔事故,以失效管段為研究對(duì)象,通過宏觀形貌分析、化學(xué)成分分析、硬度測(cè)試、金相組織分析、電鏡掃描及X射線衍射等手段,分析影響管道腐蝕的主要因素,利用電化學(xué)工作站進(jìn)行電化學(xué)試驗(yàn),明確腐蝕機(jī)理和腐蝕特征,以期為今后的腐蝕控制和延長(zhǎng)管道使用壽命提供理論依據(jù)。
1 試驗(yàn)
管段取自輸油管道靜置段腐蝕穿孔部位,通過數(shù)碼相機(jī)記錄管段內(nèi)、外壁的腐蝕情況,通過ARL-4460 直讀光譜儀、OLYMPUS GX51 型金相顯微鏡、KB 30BVZ-FA 維氏硬度計(jì)對(duì)管段的化學(xué)成分、金相顯微組織、硬度進(jìn)行分析,采用ZEISS Supra40 場(chǎng)發(fā)射電子掃描顯微鏡、EDAX XM2-60S型EDS分析儀、D/MAX-2500PC型X射線衍射儀對(duì)腐蝕產(chǎn)物進(jìn)行表征分析。
電化學(xué)試驗(yàn)在PARSTAT 273A型電化學(xué)工作站上進(jìn)行,采用經(jīng)典的三電極體系,工作電極為X65管線鋼試樣,參比電極為飽和甘汞電極,輔助電極為鉑電極。將試樣用200~1 000 目的砂紙逐級(jí)打磨,依次用無水乙醇和丙酮進(jìn)行脫脂、脫油處理,再放入烘干器中備用;腐蝕溶液采用經(jīng)脫油的采出水;電化學(xué)阻抗譜試樣施加的正弦波電位信號(hào)為10 mV,掃描范圍0.05~1×105Hz。
2 結(jié)果與討論
2.1 宏觀形貌分析
輸油管道的靜置段外壁防腐層基本完好,但腐蝕穿孔部位周圍的防腐層已脫落。采用超聲波測(cè)厚儀沿失效管段的周向進(jìn)行壁厚測(cè)量,發(fā)現(xiàn)壁厚減薄嚴(yán)重的部位多集中在4 點(diǎn)至8 點(diǎn)鐘方向,而其余方向壁厚減薄并不嚴(yán)重,12 點(diǎn)鐘方向壁厚減薄僅為0.5 mm。將管段進(jìn)行對(duì)剖后發(fā)現(xiàn),管道內(nèi)壁存在不同程度的大面積點(diǎn)蝕(圖1),最大腐蝕坑深位于6 點(diǎn)鐘方向,深度8 mm,剩余壁厚0.5 mm,最大腐蝕速率為1.52 mm/a。參照美國(guó)腐蝕學(xué)會(huì)制定的NACE RP-0775-05標(biāo)準(zhǔn),該管段屬于極嚴(yán)重腐蝕。

圖1 失效管道內(nèi)壁宏觀形貌Fig.1 Macroscopic morphology of the inner wall of failure pipeline
另外,清除管段內(nèi)存留的固體雜質(zhì)后,在對(duì)剖的管段內(nèi)壁發(fā)現(xiàn)4 條輸送介質(zhì)留下的印痕,上方2條印痕以上的管道內(nèi)壁呈黑色,下方2條印痕以下的管道內(nèi)壁呈灰白色,印痕中間為過渡帶。下方2條印痕為4 點(diǎn)鐘和8 點(diǎn)鐘方向,說明輸送介質(zhì)在該管段內(nèi)發(fā)生了油水分離現(xiàn)象,管道底部出現(xiàn)了游離水。對(duì)該站的采出水水質(zhì)進(jìn)行了檢測(cè)(表1),采出水水質(zhì)整體呈酸性,pH值較低,為5.4,沉積的游離水會(huì)溶解大量的CO2和H2S,H2S 濃度為在50.65 mg/L,同時(shí)水質(zhì)中Cl-含量較高,屬于高礦化度、高腐蝕性溶液。
綜上所述,在管道的靜置段出現(xiàn)了油水分層現(xiàn)象,4 點(diǎn)鐘至8 點(diǎn)鐘方向存在沉積的游離水,水中含有大量的腐蝕性介質(zhì),導(dǎo)致管道內(nèi)壁下部出現(xiàn)大面積腐蝕,說明該管段的腐蝕穿孔與沉積的游離水有直接關(guān)系。
2.2 化學(xué)成分分析
參照GB 4336—2002,對(duì)失效管段的外壁進(jìn)行化學(xué)成分分析,發(fā)現(xiàn)其成分符合GB/T 9711—2017中對(duì)X65 管線鋼化學(xué)成分的相關(guān)規(guī)定,結(jié)果如表2所示。

表1 采出水水質(zhì)分析結(jié)果Tab.1 Analysis results of produced water quality
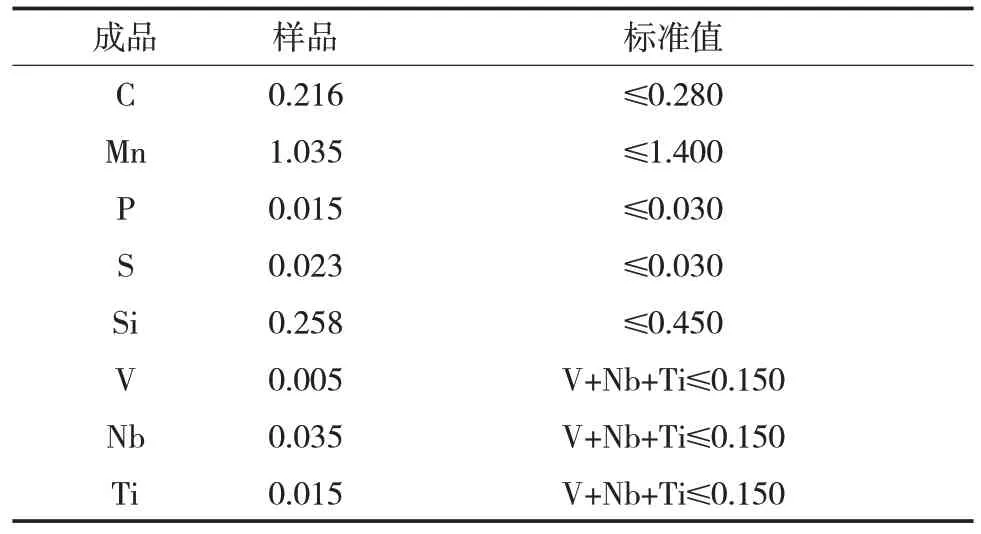
表2 失效段外壁化學(xué)成分分析Tab.2 Chemical composition analysis of the outer wall of failure part 質(zhì)量分?jǐn)?shù)/%
2.3 硬度測(cè)試
從失效管道取3 個(gè)不同位置的試樣進(jìn)行拋光后,對(duì)試樣表面進(jìn)行硬度測(cè)試,載荷10 kg,3個(gè)試樣的單點(diǎn)壓痕硬度值分別為181.6 HV10、184.3 HV10 和180.6 HV10,均符合GB/T 9711—2017 中9.10.6的相關(guān)要求(不超過345 HV10)。
2.4 金相組織分析
參照GB/T 13298—2015、GB/T 10561—2005/ISO 4967:1998 和GB/T 6394—2017 中的試驗(yàn)方法對(duì)試樣的金相組織、非金屬夾雜物以及平均晶粒度進(jìn)行分析,并采用金相顯微鏡觀察其金相組織結(jié)構(gòu),結(jié)果見表3 和圖2。分析發(fā)現(xiàn),管體金相組織為鐵素體+珠光體,組織均勻細(xì)致無異常,無MnS 和FeS等超尺寸的非金屬夾雜物。

表3 金相分析結(jié)果Tab.3 Results of metallographic analysis

圖2 失效管道金相組織結(jié)構(gòu)Fig.2 Metallographic structure of failure pipeline
2.5 腐蝕產(chǎn)物分析
對(duì)失效管道腐蝕坑內(nèi)的腐蝕產(chǎn)物進(jìn)行了電鏡掃描和EDS分析,發(fā)現(xiàn)腐蝕產(chǎn)物呈疏松多孔狀,主要的化學(xué)元素為Fe、S、C、O和Cl。為了進(jìn)一步分析腐蝕產(chǎn)物的成分,對(duì)其進(jìn)行X射線衍射,發(fā)現(xiàn)腐蝕產(chǎn)物主要為FeS 和FeCO3,還有少量的Fe3O4,無碳酸鹽和硫酸鹽(圖3、圖4、圖5)。
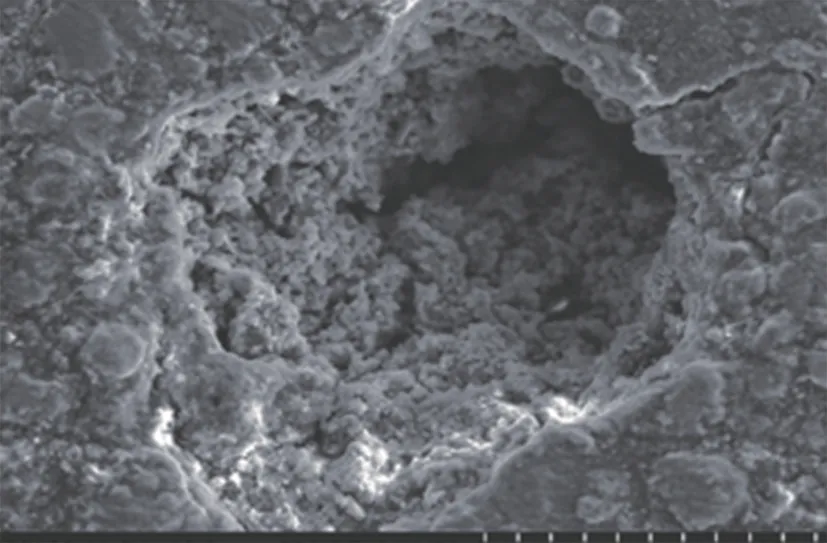
圖3 腐蝕產(chǎn)物微觀形貌Fig.3 Microstructure of corrosion products
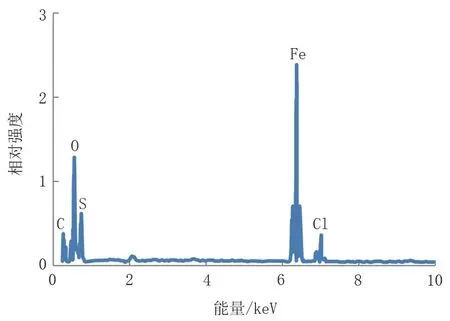
圖4 腐蝕產(chǎn)物EDS分析Fig.4 EDS analysis of corrosion products
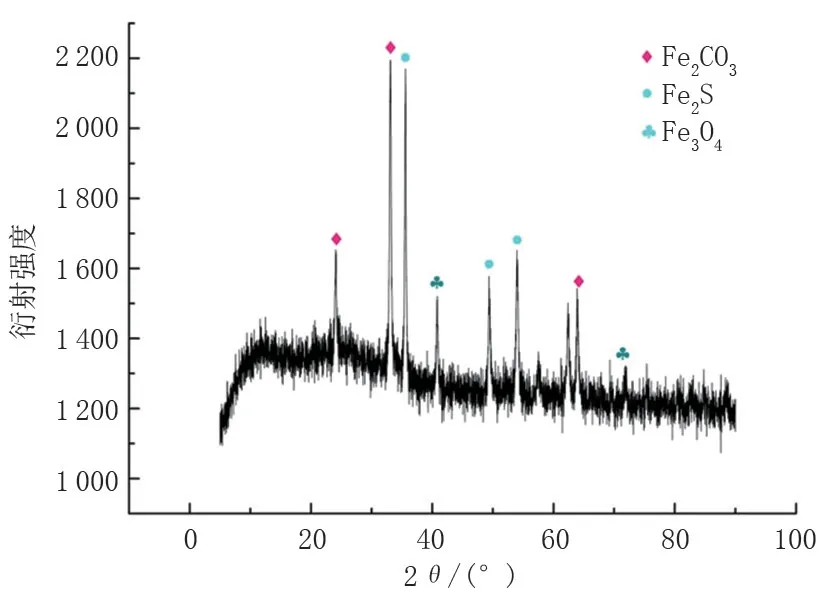
圖5 腐蝕產(chǎn)物XDR圖譜Fig.5 XDR diagram of corrosion products
2.6 腐蝕機(jī)理分析
失效管道的腐蝕主要是由于腐蝕介質(zhì)作用于管體本身引起的,因此腐蝕介質(zhì)和管體材料均構(gòu)成腐蝕的要素[6-7]。通過化學(xué)成分分析、硬度測(cè)試和金相組織分析發(fā)現(xiàn)管體本身不存在問題,而失效管道的流體流速較慢(經(jīng)測(cè)算為0.21 m/s),造成油水分層嚴(yán)重,在管道底部形成沉積的游離水,發(fā)生電化學(xué)腐蝕。從腐蝕產(chǎn)物分析來看,內(nèi)壁的腐蝕類型主要為CO2和H2S 共存環(huán)境下的酸性腐蝕。同時(shí)根據(jù)研究表明,H2S和溶解氧腐蝕不應(yīng)共存,腐蝕產(chǎn)物中氧化物可能為后期環(huán)境空氣氧化所致。
CO2溶于水后生成H2CO3,在相同的pH 值下,H2CO3的腐蝕性強(qiáng)于HCl。經(jīng)研究表明,CO2對(duì)低碳鋼的腐蝕速率高達(dá)7 mm/a[8-9];H2S極易溶于水,溶于水后發(fā)生電離反應(yīng)釋放出H+,H+作為陰極的強(qiáng)去極化劑,可以在陰極奪取電子,從而促進(jìn)陽(yáng)極的溶解反應(yīng)。此外,H+在腐蝕坑內(nèi)富集會(huì)對(duì)管材形成氫損傷,表現(xiàn)為硫化物應(yīng)力開裂、氫鼓泡等,具體的腐蝕機(jī)理如下:
H2S→H++HS
HS-→H++S2-
CO2+H2O→H2CO3→H++HCO3-
HCO3-→H++CO32-
陽(yáng)極反應(yīng):Fe→Fe2++2e-
陰極反應(yīng):2H++2e→H2
腐蝕產(chǎn)物:Fe2++S2-→FeS
Fe2++CO32-→FeCO3
腐蝕坑內(nèi)還發(fā)現(xiàn)了Cl 元素,Cl-作為催化劑,基于電價(jià)平衡原理,Cl-可以通過腐蝕產(chǎn)物膜的孔洞被優(yōu)先吸附到基材的缺陷表面,由于Cl-的不斷遷移,導(dǎo)致腐蝕坑內(nèi)的Cl-含量越來越高,金屬氯化物的水解使腐蝕坑內(nèi)的溶液進(jìn)一步酸化,生成了更多的Fe2+,從而再次促進(jìn)Cl-進(jìn)入腐蝕坑內(nèi),形成反復(fù)的水解催化反應(yīng)。此外,隨著腐蝕產(chǎn)物的不斷堆積變厚,在閉塞電池的作用下,腐蝕坑內(nèi)為缺氧狀態(tài),坑外為富氧狀態(tài),坑內(nèi)外形成氧濃差腐蝕原電池,可以進(jìn)一步破壞腐蝕產(chǎn)物膜,加速基材的腐蝕速度,使局部腐蝕全面發(fā)展,形成孔蝕破壞[10]。
2.7 電化學(xué)試驗(yàn)結(jié)果分析
通過電化學(xué)工作站對(duì)X65管線鋼試樣進(jìn)行了為期20 d 的電化學(xué)試驗(yàn),每間隔5 d 對(duì)試樣進(jìn)行電化學(xué)阻抗譜的測(cè)試。從圖6中的Nyquist圖譜可知,在去除腐蝕產(chǎn)物前,隨著試樣浸泡時(shí)間的延長(zhǎng),阻抗弧的半徑越來越小,說明腐蝕傾向越來越大。
參照ISO8407—2009 對(duì)腐蝕產(chǎn)物進(jìn)行清除,將清除腐蝕產(chǎn)物后的試樣在相同的條件下進(jìn)行電化學(xué)試驗(yàn),發(fā)現(xiàn)電化學(xué)阻抗弧的半徑較去除腐蝕產(chǎn)物前明顯變大,阻抗的實(shí)部和虛部大了一個(gè)數(shù)量級(jí),說明阻抗值增大,腐蝕傾向有所減弱(圖7)。這是由于疏松多孔的腐蝕產(chǎn)物被去除后,基材表面重新生成了一層較為致密鈍化膜,有效地保護(hù)了基材,減緩了腐蝕。同時(shí)還發(fā)現(xiàn),隨著浸泡時(shí)間的不斷延長(zhǎng),電化學(xué)阻抗弧的半徑有減小的趨勢(shì),說明腐蝕產(chǎn)物不斷堆積,繼續(xù)形成了疏松多孔的厚腐蝕產(chǎn)物,腐蝕傾向有所加強(qiáng)。因此,腐蝕產(chǎn)物的存在會(huì)導(dǎo)致腐蝕傾向更大,對(duì)整個(gè)腐蝕過程起到自加速的作用。
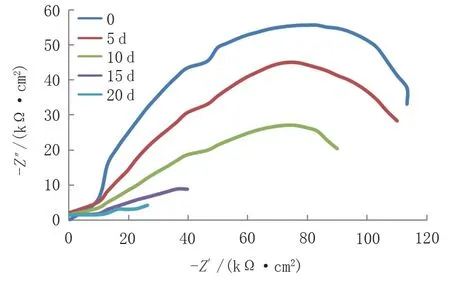
圖6 未清除腐蝕產(chǎn)物的Nyquist圖譜Fig.6 Nyquist plots of corrosion products not cleaned
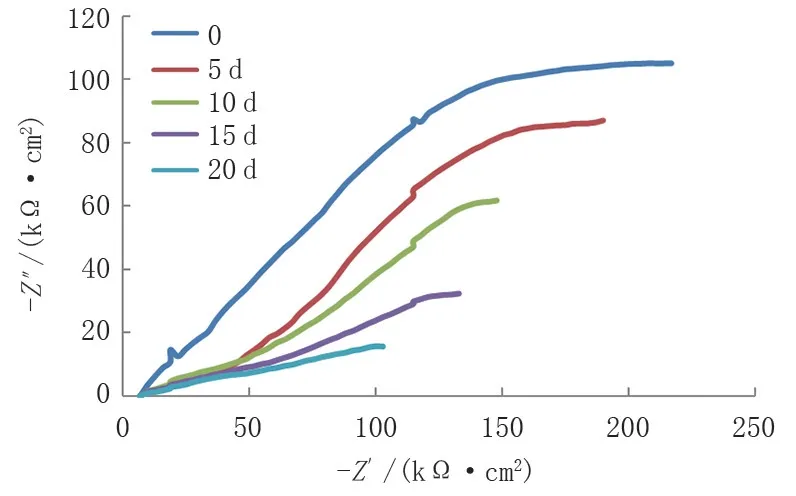
圖7 清除腐蝕產(chǎn)物的Nyquist圖譜Fig.7 Nyquist plots of corrosion products cleaned
3 結(jié)論及建議
(1)X65 管線鋼的靜置段出現(xiàn)了油水分層現(xiàn)象,4 點(diǎn)鐘至8 點(diǎn)鐘方向存在沉積的游離水,水中含有大量的腐蝕性介質(zhì),導(dǎo)致管道內(nèi)壁下部形成電化學(xué)腐蝕。因此,出現(xiàn)游離水且水中溶解有大量CO2和H2S 是腐蝕的主要原因,腐蝕類型以CO2和H2S共存環(huán)境下的酸性腐蝕為主。
(2)腐蝕機(jī)理為水解催化作用和氧濃差腐蝕原電池,腐蝕產(chǎn)物膜在一定程度上對(duì)腐蝕過程起到自加速的作用。
(3)可通過增加輸送介質(zhì)流速,降低含水率等方式避免游離水在管道底部沉積。
(4)管道靜置段的出現(xiàn)主要是由于設(shè)計(jì)和施工的錯(cuò)漏碰缺產(chǎn)生的,今后應(yīng)優(yōu)化設(shè)計(jì)和施工管理,盡量避免出現(xiàn)管道靜置段,對(duì)后期調(diào)改無法避免的部位應(yīng)加強(qiáng)腐蝕監(jiān)測(cè)。
(5)對(duì)腐蝕嚴(yán)重的重點(diǎn)部位加注緩蝕劑,可在一定程度上降低腐蝕風(fēng)險(xiǎn)。
猜你喜歡
現(xiàn)代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機(jī)設(shè)計(jì)與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:22
當(dāng)代經(jīng)濟(jì)研究(2016年5期)2016-12-01 03:12:05
現(xiàn)代農(nóng)業(yè)(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國(guó)中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學(xué)學(xué)報(bào)(社會(huì)科學(xué)版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
