穿孔素1復合雜合突變致非免疫性胎兒水腫1例并文獻回顧
2020-07-27 16:23:41童芳芳
中國當代醫藥 2020年16期
關鍵詞:檢測
童芳芳
[摘要]對穿孔素1(PRF1)復合雜合突變致非免疫性胎兒水腫臨床、實驗室及遺傳資料進行分析,檢索綜述PRF1突變與非免疫性胎兒水腫、家族性噬血細胞淋巴組織增生癥與非免疫性胎兒水腫相關文獻。在一例復發性非免疫性胎兒水腫病例發現PRF1基因新的雜合變異組合c. 03G>A(p.S168N)和c.1070G>A(p.R357Q)。文獻檢索結果顯示,PRF1致病性基因突變可致胎兒非免疫性水腫。在胎兒水腫的鑒別診斷和新生兒多器官功能衰竭的早期診斷中應考慮家族性噬血細胞淋巴組織增生癥,尤其是在找不到感染或代謝原因的情況下。噬血細胞淋巴組織增生癥的相關致病基因應納入非免疫性胎兒水腫的候選基因。本文報道1例PRF1復合雜合突變致非免疫性胎兒水腫臨床特征并對其進行病因分析。
[關鍵詞]非免疫性胎兒水腫;家族性噬血細胞淋巴組織增生癥;穿孔素1;復合雜合突變
[中圖分類號] R714.5? ? ? ? ? [文獻標識碼] A? ? ? ? ? [文章編號] 1674-4721(2020)6(a)-0190-04
[Abstract] The clinical, laboratory and genetic data of non-immune fetal edema caused by perforin 1 (PRF1) complex heterozygous mutation were analyzed. A literature review was conducted on PRF1 mutation and non-immune fetal edema, familial hemophagocytic lymphohistiocytosis and non-immune fetal edema related articles. A new heterozygous combination of PRF1 genes c.503G>A (p.S168N) and c.1070G>A (p.R357Q) was found in a recurrent non-immune fetal edema. Literature retrieving shows that PRF1 pathogenic gene mutations can cause fetal non-immune edema. Familial hemophagocytic lymphohistiocytosis should be considered in the differential diagnosis of fetal edema and early diagnosis of multiple organ failure in the newborns, especially lack of reasons for infection or metabolism. Pathogenic genes related to hemophagocytic lymphohistiocytosis should be included as candidate genes for non-immune fetal edema. This paper reported the clinical characteristics of non-immune fetal edema caused by PRF1 complex heterozygous mutation and analyzed its etiology.
[Key words] Non-immune fetal edema; Familial hemophagocytic lymphohistiocytosis; Perforin 1; Complex heterozygous mutation
胎兒水腫一般分為免疫性水腫和非免疫性水腫,是指胎兒體腔積液及軟組織水腫,表現為2處及以上的胎兒體腔異常積液,包括皮膚水腫、胸腹腔積液、心包積液,部分還會并發羊水過多或胎盤增厚[1]。免疫性水腫多發于母嬰血型不合,而非免疫性胎兒水腫由于病因復雜,被認為是一系列疾病的常見途徑,因其病因不明且致死率較高是目前胎兒醫學最為棘手的問題。
胎兒宮內感染、血液、淋巴系統疾病、先天性結構異常、胎盤臍帶因素、心血管疾病、遺傳代謝性疾病如黏多糖病、戈謝病、小兒半乳糖血癥、唾液酸貯積癥等以及先天性糖基化障礙均能引起胎兒非免疫性水腫[2-3]。在地中海貧血高發區域,由珠蛋白變異引起的巴氏水腫目前是病因明確并能夠通過產前篩查和產前診斷實現防控的單基因遺傳病。非免疫性胎兒水腫的病理生理原因復雜多樣,其診斷目前主要依賴于超聲影像學檢查,整體預后不良,明確病因對于指導宮內治療和優生遺傳具有非常重要的意義。
近年來隨著分子診斷技術的發展,關于非免疫性胎兒水腫的遺傳學研究也有了很多突破性的發現。規范并細化非免疫性胎兒水腫的診療流程,加強產前傳染性疾病的檢測和代謝病檢測,對病因不明的非免疫性胎兒水腫提倡進行基因組拷貝數變異的檢測(CNVs)甚至高通量測序以明確其遺傳學病因。
家族性噬血細胞淋巴組織細胞增生癥(familial hemophagocytic lymphohistiocytosis,FHL)是一種常染色體隱性遺傳病,大多數患有FHL的兒童在出生后2~6個月會發病,在新生兒中罕見,經常被完全忽略或者只在尸檢中被診斷出來。胎發性FHL更為罕見,被認為是FHL最嚴重的形式。目前已經明確與FHL發病相關的4個免疫缺陷基因:穿孔素1(perforin 1,PRF1)(FHL2),UNC13D(FHL3),STX11(FHL4)和STXBP2(FHL5),其組織學特征表現為細胞毒性T淋巴細胞和自然殺傷細胞的過度增生與活化,產生大量細胞因子[4]。FHL在胚胎期可表現為胎兒水腫[5-7]。本文報道1例PRF1復合雜合突變致非免疫性胎兒水腫,并對相關文獻進行回顧。
1病例資料
孕婦孕2產0;家族史:父母體健,非近親婚配,母孕期無放射接觸史。患者自訴2014年第1次懷孕于23+3周于外院B超診斷胎兒皮下水腫并引產,未進行胎兒病理學檢測。2016年10月2日,孕23+5周超聲系統篩查:單活胎,胎兒全身皮膚增厚,鼻骨顯示不清(鼻骨發育不良或缺如?),上嘴唇增厚,回聲低,小下頜,雙肺體積小,雙側胸腔積液,胎兒雙手姿勢異常,雙下肢姿勢異常,雙足馬蹄內翻可能,羊水多。2016年10月13日因停經25+1周,胎兒先天畸形要求入院行流產術。
夫妻雙方血型均為O,RH陽性,不規則抗體陰性;夫妻二人常規外周血染色體核型分析正常,血紅蛋白電泳正常;孕16+4周唐氏血清學篩查低風險。孕婦白細胞:10.9×109/L,紅細胞:3.12×1012/L,血紅蛋白99 g/L;肝腎功能檢測:總蛋白55.5 g/L;空腹血糖、凝血功能、尿常規無明顯異常;甲狀腺功能正常,常規傳染病檢測(乙肝、丙肝、艾滋、梅毒、甲肝、戊肝)均為陰性;葡萄糖-6磷酸脫氫酶正常,糖耐量實驗正常。2016年10月16日經陰道排出一死女嬰。
采用Affymetrix CytoScan HD芯片對流產胎兒組織進行全基因組掃描,檢測前流產物DNA經母血DNA連鎖分析排除母體細胞污染。整套染色體未檢出與臨床致病性相關的基因拷貝數變化和大片段純合子。
對流產胚胎皮膚組織DNA及父母乙二胺四乙酸(EDTA)的抗凝外周血,送往廣州嘉檢醫學檢驗中心針對臨床相關的4000個致病基因進行檢測和分析,檢測結果質控統計:檢測區間包括4000個相關基因,50 584個編碼區總共含有8 421 879個堿基。平均覆蓋深度268+/-167X,>10X覆蓋區間占98.8%,>20X覆蓋區間占98.3%。PRF1基因兩個雜合變異c.503G>A(p.S168N)和c.1070G>A(p.R357Q),測序數據顯示這兩個變異分別遺傳自送檢者的父親和母親(均為雜合狀態)。
這兩個突變在SNP數據庫中有記載,頻率<1%,所在區域是這個蛋白質的重要組成部分,不同物種的氨基酸序列高度保守。經計算機輔助分析和預測,這兩個變異均有可能對相應蛋白質結構/功能產生影響。結合送檢者的臨床表現和家系分析,依據美國ACMGG變異分類指南,這兩個變異暫定為“Ⅱ類可能致病”。PRF1 c.503G>A變異在淋巴組織細胞增生癥相關的臨床病例中已經有報道[8]。
進一步隨訪,2017年于中信湘雅生殖遺傳專科醫院經過遺傳咨詢和家系驗證并通過PGD于2019年4月7日選擇移入兩個PRF1雜合突變胚胎,正常產檢,孕18+5周羊水穿刺基因檢測胎兒PRF1 c.503G>A(p.S168N),c.1070位點未見異常,28周大排畸胎兒發育正常。
2討論
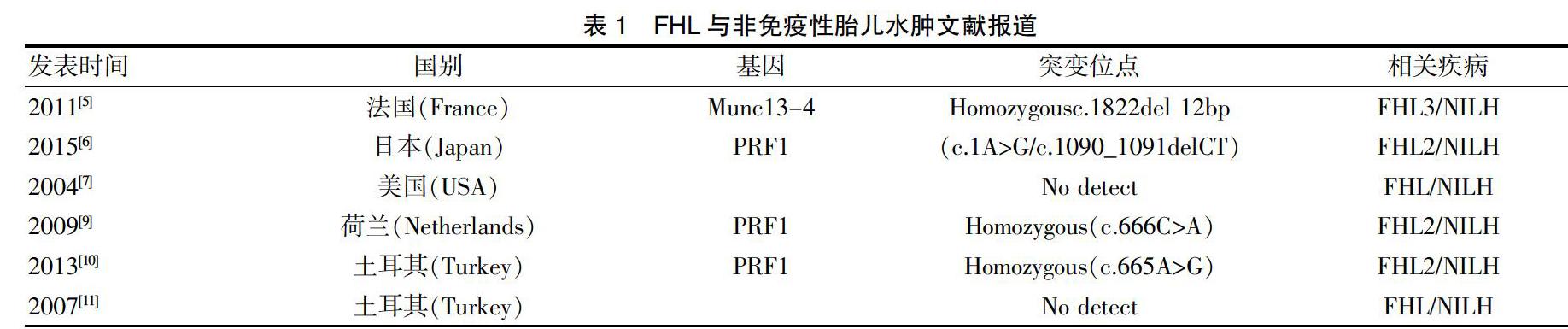
以“non-immune hydrops fetalis AND PRF1”和“familial hemophagocytic lymphohistiocytosis AND non-immune hydrops fetalis”為檢索式檢索PubMed數據庫,以全文“噬血淋巴組織增生癥并含非免疫性胎兒水腫”或者全文“PRF1并含非免疫性胎兒水腫”為檢索式模糊檢索萬方數據庫和中國知網,檢索時間均為建庫至2019年10月20日,共檢索到英文文獻6篇詳細報道了非免疫性胎兒水腫與噬血淋巴組織增生癥的臨床關聯,現將相關病例信息在表1中列出。
PRF1基因定位于人類染色體10q22.1,編碼孔形成蛋白,是一種儲存于細胞毒性T淋巴細胞和自然殺傷細胞內囊泡胞質顆粒中高度保守的糖蛋白,是膜攻擊復合物/穿孔素超家族成員之一,是人類免疫系統殺傷和清除靶細胞的重要效應分子[12]。穿孔素通過在靶細胞膜上形成孔來誘導細胞毒顆粒進入靶細胞胞質從而消除病毒和其他因素感染和轉化的細胞,基因突變導致蛋白表達缺失或功能不全,使細胞毒性T淋巴細胞和自然殺傷細胞細胞毒性功能受損,不能誘導靶細胞凋亡和清除,抗原提呈細胞過度且持續活化,組織細胞和T淋巴細胞導致全身性炎癥以及臨床表現,在臨床上主要表現為生長發育不良、黃疸、肝脾腫大、全血細胞減少并有神經系統表現,如癲癇、共濟失調。該基因的突變與多種人類疾病相關,包括糖尿病、多發性硬化癥、淋巴瘤、自身免疫性淋巴增殖性綜合征(ALPS)、再生障礙性貧血、FHL2[13-14]。
Vermeulen等[9]經分子診斷證實PRF1 c.666C>A(p.His222Gln)純合突變導致的FHL引起胎兒水腫。先證者姐弟同為純合突變引發FHL,一個在產前出現,另一個在新生兒時出現。前一例患兒死于產前胎兒水腫,胎兒內部檢查顯示皮下水腫、心包、胸腔積液及腹腔積液,許多單核細胞在血管內被發現,部分伴有紅細胞吞噬。而第二例患兒出生后不久出現致命的多器官衰竭,尸檢顯示嚴重的皮下水腫、腹腔積液、胸膜及心包積液。PRF1 c.665A>G(p.His222Arg)純合突變噬血細胞性淋巴組織細胞增多癥致非免疫性胎兒水腫在另一同卵雙胞胎中進一步證實[15]。
迄今為止已經報道穿孔蛋白致病突變已超過115個,分布在其整個功能領域,沒有明顯的聚類。但是非免疫性胎兒水腫和該基因突變的分子遺傳學關聯分析還比較少,2004年Malloy等[7]將非免疫性胎兒水腫與噬血淋巴組織增生癥關聯起來,孕32周孕婦羊水多,雙胎之一腹腔積液,胸腔積液,心包積液32周剖宮產新生兒全身水腫,雙側胸腔積液,排除病毒和感染因素,病理學和解剖學是各項指標符合HLH診療標準,但是由于條件有限沒有在分子診斷水平進行驗證。隨著高通量測序技術在臨床上的廣泛應用,因為一些特殊的病例,如復發性非免疫性胎兒水腫和同卵雙胎純合突變胎兒的臨床表現使研究人員逐漸確認這種通常在新生兒和嬰幼兒中發病率較高的疾病在胎兒期的臨床表現更為劇烈。
噬血細胞淋巴組織增生癥在臨床表現出高度的遺傳異質性,患者年齡從胎兒到60歲老人,呈現出不同的一般是外源性觸發和遺傳易感性共同作用的結果。在這個組合中,外源性和遺傳因素的不同權重解釋了從繼發性到嚴重感染到家族性聚集的廣泛疾病譜[16-17]。
本病例中,可能存在的誘發原因由于一些實際因素還沒有進行系統而全面的篩查。因此在實驗室建立和健全病原學、細胞學及相關酶學和基因檢測對于非免疫性胎兒水腫病因的分析至關重要。
在胎兒非免疫性病因引起水腫的鑒別診斷和新生兒多器官功能衰竭的早期診斷中,尤如果找不到感染或代謝原因,應考慮FHL的可能。
[參考文獻]
[1]侯磊,王欣.非免疫性胎兒水腫的診療新進展——2018年《非免疫性胎兒水腫的調查和管理指南》解讀[J].中國全科醫學,2018,21(35):4289-4294.
[2]Makhamreh MM,Cottingham N,Ferreira CR,et al.Nonimmune hydrops fetalis and congenital disorders of glycosylation:A systematic literature review[J].J Inherit Metab Dis,2019.Epub ahead of print
[3]麻希洋,吳慶華.非免疫性胎兒水腫的遺傳學研究進展[J].中華醫學遺傳學雜志,2018,35(1):125-128.
[4]Meeths M,Chiang SC,Lfstedt A,et al.Pathophysiology and spectrum of diseases caused by defects in lymphocyte cytotoxicity[J].Exp Cell Res,2014,325(1):10-17.
[5]Bechara E,Dijoud F,De Saint Basile G,et al.Hemophagocytic lymphohistiocytosis with Munc13-4 mutation:a cause of recurrent fatal hydrops fetalis[J].Pediatrics,2011,128(1):e251-e254.
[6]Iwatani S,Uemura K,Mizobuchi M,et al.Familial hemophagocytic lymphohistiocytosis presenting as hydrops fetalis[J].AJP Rep,2015,5(1):e22-e24.
[7]Malloy CA,Polinski C,Alkan S,et al.Hemophagocytic lymphohistiocytosis presenting with nonimmune hydrops fetalis[J].J Perinatol,2004,24(7):458-460.
[8]Lu G,Xie ZD,Shen KL,et al.Mutations in the perforin gene in children with hemophagocytic lymphohistiocytosis[J].Chin Med J(Engl),2009,122:2851-2855.
[9]Vermeulen MJ,De Haas V,Mulder MF,et al.Hydrops fetalis and early neonatal multiple organ failure in familial hemophagocytic lymphohistiocytosis[J].Eur J Med Genet,2009,52(6):417-420.
[10]Balta G,Topcuoglu S,Gursoy T,et al.Association of nonimmune hydrops fetalis with familial hemophagocytic lymphohistiocytosis in identical twin neonates with perforin His222Arg (c665A>G) mutation[J].J Pediatr Hematol Oncol,2013,35(8):e332-e334.
[11]Yetgin S,Aytac S,Gurakan F.Nonimmune hydrops fetalis in two cases of consanguineous parents and associated with hereditary spherocytosis and hemophagocytic hystiocytosis[J].J Perinatol,2007,27(4):252-254.
[12]Law RH,Lukoyanova N,Voskoboinik I,et al.The structural basis for membrane binding and pore formation by lymphocyte perforin[J].Nature,2010,468(7322):447-451.
[13]Stepp SE,Dufourcq-Lagelouse R,Le Deist F,et al.Perforin gene defects in familial hemophagocytic lymphohistiocytosis[J].Science,1999,286(5446):1957-1959.
[14]Fink TM,Zimmer M,Weitz S,et al.Human perforin (PRF1) maps to 10q22,a region that is syntenic with mouse chromosome 10[J].Genomics,1992,13(4):1300-1302.
[15]An O,Gursoy A,Gurgey A,et al.Structural and functionalanalysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients[J].Protein Sci,2013,22(6):823-839.
[16]Mhatre S,Madkaikar M,Desai M.Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis(FHL)patients in India[J].Blood cells Mol Dis,2015,54(3):250-257.
[17]Gao L,Dang X, Huang L,et al.Search for the potential"second-hit" mechanism underlying the onset of familial hemophagocytic lymphohistiocytosis type 2 by whole-exome sequencing analysis[J].Transl Res,2016,170:26-39.
(收稿日期:2019-10-23? 本文編輯:任秀蘭)
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
