介孔NiS2/S-g-C3N4的制備及其光催化產氫性能研究*
2020-08-03 03:29:10蘇揚航韓亞翔陳緒興
功能材料 2020年7期
關鍵詞:催化劑
劉 凱,蘇揚航,韓亞翔,陳緒興,高 云,李 榮,2
(1. 湖北大學 材料科學與工程學院, 武漢 430062; 2. 結構化學國家重點實驗室, 福州 350002)
0 引 言
隨著人類社會的快速發展,以化石能源的枯竭所帶來的能源問題日益凸顯,利用半導體光催化分解水產氫被認為是最有希望解決能源危機的技術之一。1972年Fujiashima等人發現TiO2可以光電催化分解水制氫,從此拉開了光催化分解水制氫的序幕[1]。隨后針對TiO2光催化分解水制氫活性的優化,科研工作者展開了大量研究[2-8]。由于TiO2較大的禁帶寬度(3.2 eV)使其僅對太陽光譜中的紫外光具有響應,而紫外光僅占太陽光譜中能量約5%,導致其光能利用率低,實際應用受到了極大限制[9]。2009年,福州大學王心晨教授發現石墨烯相氮化碳(g-C3N4)在可見光下可以分解水制氫[10],這種化學性質穩定,易于制備、成本低廉的具有類石墨烯層狀結構的材料在光催化領域迅速引起了廣泛的關注[11]。g-C3N4的禁帶寬度約為2.7 eV,可以吸收太陽光譜中λ≤460 nm的藍紫光,相較于傳統的金屬氧化物半導體光催化劑,其光能利用率較高,然而較低的比表面積、較高的載流子復合率及較低的表面反應速率[12-14],導致純g-C3N4表現出非常低的光催化產氫性能,嚴重制約了g-C3N4的實際應用。
針對上述問題,科研工作者展開了大量研究。對g-C3N4進行摻雜,以調節g-C3N4的光學、電子能帶結構等特性。例如:2009年,王心晨教授等報道在g-C3N4框架中摻雜金屬元素Zn2+和Fe2+,并且發現隨著摻雜金屬元素含量的增加,金屬/g-C3N4復合材料相比g-C3N4擴大了其對太陽光譜的光吸收范圍[15]。2010年,成會明教授等制備了S摻雜g-C3N4,優化了催化劑的電子結構,價帶的負移增加了光生空穴的氧化能力[16]。2011年,王心晨教授等又發現除了Zn2+、Fe2+離子外,其他過渡金屬陽離子Ni2+、Cu2+、Co3+和Mn3+等,在不破壞g-C3N4主體結構的前提下,都可以摻入g-C3N4的框架中,并且表現出相似的效應[17]。2012年,陳亦琳教授等通過簡單的H2O2水熱處理合成了O摻雜g-C3N4,吸收帶邊移動至498 nm,有效地增強了可見光響應[18]。綜上所述,元素摻雜可以明顯拓展g-C3N4的吸收光譜。理論上單層g-C3N4具有較大的比表面積,然而通常制備的塊狀g-C3N4存在嚴重的層堆積現象,導致其比表面積較低,光生電子和空穴還沒有遷移到催化劑表面參與反應就在催化劑體內復合,載流子復合率較高。針對這個問題,研究者采用剝離法來制備較大的比表面積的g-C3N4納米片。例如:2012年,成會明教授等在空氣中對塊狀g-C3N4進行熱氧化剝離,獲得了厚度約為2 nm,比表面積為306 m2/g的g-C3N4納米片,其電子傳輸能力提高并且載流子壽命增加,光催化產氫性能得到了提高[19]。2013年,王心晨教授等通過簡單的液相超聲剝離塊狀g-C3N4得到g-C3N4的納米片,納米片的厚度約為2 nm,比表面積更是高達384 m2/g,在可見光下其光催化產氫性能提高了大約9倍[20]。同時,將兩種具有合適導帶和價帶位置的報道提構成異質結,利用異質結的內建電場來促進電荷轉移和分離,也是一種有效的方法。例如:2011年,朱永法教授等制備了ZnO/g-C3N4雜化納米材料,在紫外線照射下光催化活性增加了3.5倍[21]。2011年,John T.S. Irvine教授等發現SrTiO3-C3N4復合光催化劑在可見光產氫速率達到440 μmol/(h·g),純g-C3N4的光催化性能提高很多倍[22]。盡管通過剝離、構建異質結可以促進光生載流子分離,但是g-C3N4表面較低的反應速率又大大抑制了光催化反應速率,負載助催化劑也是一種行之有效的方法,例如:2009年,王心晨教授等用光化學沉積的方法,在g-C3N4上沉積不同的貴金屬納米顆粒(Rh、Ru、Pd、Pt、Au等),并比較了其在可見光下分解水制氫的性能,發現Pt是最好產氫助催化劑,光催化產氫效率提高了7倍[23]。2010年,Markus Antonietti教授等通過沉積-沉淀、光沉積和浸漬等方法在g-C3N4上沉積金納米顆粒,使其光催化活性得到明顯提升[24]。然而由于貴金屬的昂貴價格,導致其不可能進行大規模使用。過渡金屬硫化物由于其獨特的光學和電學性質[25],H2在過渡金屬硫化物表面的吸附自由能接近于零[26],被認為是一種理想的產氫助催化劑,可有效的提高g-C3N4的光催化產氫活性。例如:2013年,王心晨教授等在g-C3N4中引入了具有良好電催化產氫活性的MoS2,顯著提高其光催化活性[27]。2014年,方曉明教授等通過一種簡單的原位無模板離子交換工藝制備了NiS/g-C3N4復合光催化劑,并在光催化析氫測試中,最佳性能的樣品g-C3N4/NiS-1.5%(摩爾分數)與2.0%(質量分數)Pt/g-C3N4的產氫速率幾乎接近,NiS作為助催化劑可以有效的促進g-C3N4光生載流子的分離,從而提高光催化產氫活性[28]。
目前,經過科研工作者的努力,g-C3N4光催化產氫性能得到了顯著提升,然而上述針對g-C3N4光催化活性的改善方法依然還存在一些缺點。例如:剝離制備的g-C3N4雖然具有超大的比表面積,但是其制備方法的產量極低,并不利于大規模制備。所制備的異質結界面接觸不夠良好,導致光生載流子分離效率依然有待進一步提高。同時,僅僅采用元素摻雜、形貌調控、構造異質結和負載助催化劑等改性方法中的一種或是兩種方法的結合,其改善能力有限。如何采用一種經濟、簡單的制備方法將上述所有改善方法集于一體以進一步提升g-C3N4的光催化產氫性能依然是一個挑戰。
鑒于上述考慮,本文中我們利用介孔SiO2作為硬模板,采用CH4N2S和Ni(CH4N2S)4作為前驅體,通過一步退火法成功制備了介孔S-g-C3N4負載NiS2復合光催化劑。利用介孔材料的有效傳質效應;同時利用Ni-S配位原子層作為S-g-C3N4和NiS2的過渡連接層,確保了S-g-C3N4和NiS2間光生載流子的高效傳輸;最后利用非貴金屬NiS2作為助催化劑來降低析氫過電位,促進H+的電催化還原,這為獲得高效的g-C3N4基光催化產氫材料提高了保障,使其在可見光下(λ≥420 nm)的產氫性能達到141.33 μmol/(g·h),是純g-C3N4的46倍。然后通過XPS、穩態/瞬態熒光光譜、電化學阻抗譜、線性伏安曲線、瞬態光電流對其光催化活性提升機理進行了進一步探究。
1 實 驗
1.1 x-NiSCN復合光催化劑的制備
以CH4N2S和Ni(CH4N2S)4為前驅體,通過熱縮聚的方法原位制備了具有不同NiS2含量的x-NiSCN納米復合材料。具體實驗步驟如下:首先,將5 g的粒徑約為20 nm的SiO2納米微球分散于100 mL去離子水中,在劇烈攪拌下加入10 g硫脲和一定比例的四水合乙酸鎳,并攪拌30 min。其次,將所得溶液加熱至100 ℃進行蒸發。待水分蒸干后將所得到的固體粉末在60 ℃干燥12 h,并進行充分研磨。再次,將研磨后的固體粉末置于帶蓋的剛玉坩堝中,在N2氛圍下525 ℃退火,保溫2 h,升溫速率為5 ℃/min。最后,將退火后的樣品分散在2M NaOH溶液中以刻蝕掉SiO2模板,并通過離心和抽濾洗滌得到最終樣品,將其標記為x-NiSCN,其中x代表硫脲和乙酸鎳的質量比。采用上述相同的制備方法,分別以采用尿素和硫脲作為原料,但不加入四水合乙酸鎳制備了g-C3N4(標記為g-C3N4)和S摻雜的g-C3N4(標記為SCN)樣品。
1.2 催化劑的表征
通過粉末X射線衍射儀(D8-Advance,X射線源Cu靶Kα射線,λ=0.15418 nm)和傅立葉紅外光譜儀(Spectrum One)表征樣品的物相結構于表面基團;使用分光光度計(日本SHIMADZU UV-3600)測試樣品的U紫外-可見漫反射吸收光譜(UV-Visible DRS);使用FESEM(蔡司SIGMA 500)、TEM(Tecnai G20)觀察樣品的形態;利用X射線光電子能譜(XPS,ESCALAB 250Xi)對樣品的組成及化學態進行表征,利用樣品表面上污染碳的284.8 eV C 1s峰對所有結合能進行校準;利用熒光光譜儀(Perkin Elmer LS55)測試樣品的瞬態/穩態吸收光譜,采用375 nm氙燈作為激發光源。
1.3 光催化產氫測試
光催化產氫實驗采用北京中教金源的CEL-SPH2N光催化活性評價系統進行測試。采用超聲法將10 mg光催化劑分散在50 mL水溶液(20 vol%三乙醇胺)中,加裝420 nm濾光片的300 W氙燈作為光源,每隔60 min進行一次取樣并利用氣相色譜儀(GC7920,以N2為載氣,TCD)分析H2的生成量。
1.4 光電化學測試
使用辰華電化學工作站(CHI 600E)的三電極系統進行光電化學測試,以Pt絲電極作為對電極,Ag/AgCl電極作為參比電極,在導電表面上涂有樣品的FTO玻璃作為工作電極。工作電極制備過程如下:首先,將10 mg樣品分散在0.5 mL DMF中,將其充分超聲分散;然后,滴到FTO玻璃的導電表面上,最后,將獲得的工作電極在50 ℃下加熱約3 h,以蒸發殘留的溶劑。光源為300 W氙燈,電解液為0.5 mol/L Na2SO4水溶液。在瞬態光電流響應和電化學阻抗譜(EIS)測試中。在0.1~106Hz的頻率范圍內以5 mV的振幅記錄EIS譜線。在線性掃描伏安法(LSV)測量中,掃描范圍為-0.65~-1.35 V(相對于Ag/AgCl,pH=6.6)。
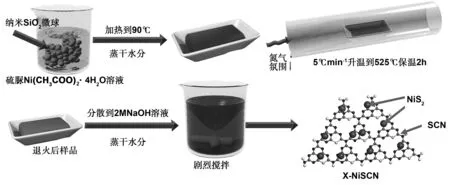
圖1 x-NiSCN復合材料制備示意圖Fig 1 Schematic illustration for the fabrication of x-NiSCN composites
2 結果及討論
2.1 形態和微觀結構
所制備樣品的形貌如圖2所示。從圖2(a)的SEM可知,SCN樣品為褶皺的片狀,圖2 (c)的TEM圖進一步顯示該片層的厚度為納米級。從圖2(b)可知,在負載NiS2后,100-NiSCN樣品仍然保持褶皺的片狀,并且表面沒有發現明顯的NiS2大顆粒,圖2 (d)的TEM圖進一步顯示NiS2納米顆粒的尺寸大約為20 nm左右,并且較為均勻負載在SCN片上。

圖2 SCN和100-NiSCN的SEM和TEM圖Fig 2 SEM and TEM images of SCN and 100-NiSCN
2.2 物相結構與表面化學狀態
所制備樣品的XRD圖譜如圖3 (a)所示,g-C3N4和SCN都在2q為12.7和27.3°位置出現了明顯的衍射峰,分別對應g-C3N4的(100)和(002)晶面。隨著乙酸鎳添加量的增加,SCN特征峰強度逐漸降低但并沒有消失,說明其結構依然得以保存,而NiS2的特征峰的強度逐漸增強,并且隨著乙酸鎳含量的增加,特征峰的強度逐漸增加。對其基團進一步進行了FT-IR表征,如圖3 (b)所示。g-C3N4,SCN和x-NiSCN均出現了位于1 100~1 700 cm-1處的CN雜環的特征峰和815 cm-1處的s-三嗪單元的尖銳特征峰,以及3 000~3 600 cm-1處的N-H特征寬峰。除此之外,在x-NiSCN中并沒有觀察到其他的特征峰,進一步說明NiS2的負載并沒有破壞g-C3N4的本征結構。

圖3 g-C3N4、SCN和x-NiSCN的XRD圖和(b) FT-IR圖譜Fig 3 XRD patterns and FT-IR spectrum of g-C3N4, SCN and x-NiSCN
通過X射線光電子能譜(XPS)研究了樣品的表面化學性質。所有圖譜的結合能都利用284.8 eV處出現的污染碳源的C 1s峰進行校準。圖4 (a)是NiS2、SCN和100-NiSCN的C 1s高分辨圖譜,NiS2中只含一個位于284.8 eV的峰,歸屬于表面污染碳的C-C鍵。在SCN樣品中,碳有3個峰,分別位于284.8、286.3和288.0 eV,分別對應于碳C—C鍵、S摻雜到g-C3N4結構中的C—S鍵和g—C3N4中三嗪的N—CN2結構中的碳。100-NiSCN的C 1s譜圖類似于SCN,但是N—CN2結構中的碳結合能向高移到288.2 eV,而C-S鍵的結合能向低移到285.9 eV。圖4 (b)是SCN和100-NiSCN的N 1s高分辨圖譜,對于SCN,N 1s圖譜有4個峰,分別位于398.3、400.1、401.1和404.3 eV,它們分別是CN—C、(C)3-N、N-H中的N以及N的π激發。在100-NiSCN的中發現CN—C和N的π激發分別向高結合能移動到398.7和404.8 eV,而(C)3—N、N—H則向低移動至399.4和400.7 eV。圖4(c)是NiS2,SCN和100-NiSCN的S 2p的高分辨圖譜。SCN樣品中的兩個峰,分別位于162.3和167.8 eV,它們分別是由硫摻雜的g-C3N4材料中的硫取代氮而形成的C—S鍵以及材料表面的S-O鍵。NiS2中的3個峰,位于162.8和164.0 eV的是與Ni2+離子形成二硫化物的還有在168.4 eV為NiS2表面S-O鍵。在100-NiSCN樣品中值得注意的是,Ni-S中的兩個峰都向低結合能移動到162.5和163.7 eV,而在g-C3N4中摻雜的C-S鍵向高結合能移動到164.3 eV。圖4 (d)是NiS2和100-NiSCN的Ni 2p的高分辨圖譜。在NiS2中,854.0和871.4 eV分別是Ni 2p 3/2和Ni 2p 1/2,顯示為Ni2+狀態,857.8和877.9 eV為Ni的衛星峰。而在100-NiSCN中,所有的峰都向高結合能方向移動,Ni 2p 3/2和Ni 2p 1/2移動至855.7和873.4 eV,衛星峰移動至862.1和879.5 eV。

圖4 SCN、NiS2和100-NiSCN的XPS高分辨能譜Fig 4 High-resolution XPS spectra of SCN, NiS2 and 100-NiSCN
2.3 光學吸收
通過紫外-可見漫反射吸收光譜來研究樣品的光吸收特性,如圖5(a)所示。利用硫脲作為前驅體制備的SCN的吸收帶邊,相對于g-C3N4吸收帶邊出現了的紅移,禁帶寬度進一步較小,從而提升了太陽光譜的利用率。具有不同NiS2含量的x-NiSCN復合樣品均顯出類似SCN的吸收邊,這表明SCN的結構得以保存。但是x-NiSCN復合樣品在可見光區域(500~800 nm)的吸光度大大提高,并且隨著NiS2含量的增加,吸光度逐漸增大,這可以歸因于黑色的NiS2較寬的光譜吸收。
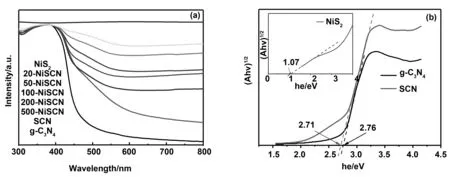
圖5 g-C3N4,SCN,NiS2和x-NiSCN樣品的紫外-可見漫反射吸收光譜和SCN和NiS2對應的Kubelka-Munk圖Fig 5 UV-visible DRS of g-C3N4, SCN, NiS2 and x-NiSCN and Kubelka-Munk plots of SCN and NiS2
2.4 光催化產氫性能
g-C3N4、SCN和x-NiSCN的光催化產氫性能如圖6所示。正如前言所述,由于較高的載流子復合效率和滯緩的表面反應速率,g-C3N4表現出微弱的光催化產氫性能,其產氫速率僅為3.05 μmol/(g·h)。雖然S元素的摻雜使SCN在可見光區的吸收增加,導致其光催化產氫速率提升到了5.21 μmol/(g·h),但由于其滯緩的反應動力學導致光催化產氫性能依然較低。在負載了NiS2之后,其光催化產氫性能得到了顯著的提升,其中100-NiSCN樣品表現出最高的光催化產氫性能,其產氫速率為141.33 μmol/(g·h),分別為g-C3N4和SCN的46和27倍。結果表明,S元素摻雜和助催化劑NiS2的引入可以有效的提高g-C3N4的光催化產氫性能。

圖6 g-C3N4、SCN和x-NiSCN復合材料的光催化產氫性能Fig 6 Photocatalytic hydrogen production performance of g-C3N4, SCN and x-NiSCN
2.5 光催化機理研究
為了進一步揭示光催化產氫性能提升的原因,對所制備樣品進行了穩態/瞬態熒光光譜、電化學阻抗譜、線性伏安和瞬態光電流表征。如圖7(a)所示,與之前報道一樣,SCN樣品在400~600 nm處有一個較寬的發射峰,為帶間躍遷發射峰。在100-NiSCN的樣品中,由于NiS2的引入,導致其發光光譜強度明顯減弱,從而說明負載NiS2可顯著降低光生載流子的復合效率。在圖7 (b)的瞬態熒光光譜表征中,SCN樣品光生載流子的平均壽命為1.54 ns,而負載NiS2之后,其光生載流子的平均壽命提升到1.80 ns,進一步證實了負載NiS2可促進光生電子-空穴的分離效率。從圖7(c)所示的電化學阻抗譜中,可知SCN樣品具有較大的電阻,而NiS2具有良好的導電性,隨著NiS2的引入,SCN的導電性得到明顯的改善,這有利于光生載流子的傳輸。從圖7(d)中的線性伏安曲線可知,對于電催化析氫反應,SCN具有較大的過電勢,這是由于其滯緩的反應動力學所致。而NiS2表現出良好的電催化析氫性能,可見其為一種良好的產氫助催化劑。因此,在引入NiS2后,SCN的電催化析氫性能得到了明顯改善。上述實驗結果表明NiS2的引入可顯著提升SCN中光生電子與空穴的分離、運輸,并有效改善其滯緩的反應動力學,從而可預測NiS2的引入可有效提高其光電催化性能。我們利用瞬態光電流進行了驗證,如圖8所示,相比于單純的SCN和NiS2樣品,100-NiSCN樣品的光電流得到了顯著提升,從而證實了上述預測。
通過上述結構表征及機理研究,可知100-NiSCN具有良好的光催化產氫性能的主要原因是:(1)利用SiO2作為硬模板合成介孔材料,提升了催化劑的傳質能力;(2)S元素的摻雜拓展了g-C3N4對太陽光譜的吸收;(3)NiS2與SCN良好的界面接觸促進了光生載流子的分離;(4)助催化劑NiS2的引入改善了g-C3N4本身滯緩的反應動力學,降低了其產氫過電位。
3 結 論
利用納米SiO2作為硬模板,采用CH4N2S和Ni(CH4N2S)4作為前驅體,通過簡單的退火法成功制備了介孔S-g-C3N4負載NiS2復合光催化劑。在可見光下 (λ≥420 nm),100-NiSCN樣品的產氫速率可達141.33 μmol/(g·h),是純g-C3N4的46倍。然后我們利用XRD、SEM、TEM、UV-visble吸收光譜、FT-IR、XPS、穩態/瞬態熒光光譜、電化學阻抗譜、線性伏安曲線、瞬態光電流對其結構與形貌、表面狀態、光催化反應機理進行了詳細研究,揭示了其光催化產氫性能得以提升的原因。該研究為制備高效的g-C3N4基光催化劑提供一種簡單、高效、經濟的設計思路,并為其進行大規模工業化生產及應用提供了理論基礎。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
