基于檸檬酸體系的中藥電化學指紋圖譜的應用研究△
2020-08-21 06:32:58杜寶中王昊陽樊花余中劉廣鈞
中國現代中藥 2020年6期
杜寶中,王昊陽,樊花,余中,劉廣鈞
西安理工大學 應用化學系,陜西 西安 710054
中藥以其純天然性、毒副作用低、標本皆治等特點,數千年來為中華民族的健康和繁衍做出了巨大貢獻,被譽為中華民族的寶貴文化遺產;但因中藥組成的復雜性及藥效的多組分協同效應,導致其質量的一致性和臨床療效的穩定性受到較大影響,從而成為制約中藥的現代化進程和“鋪軌”國際市場關鍵所在。因此,如何根據中藥的特點,對其化學成分及含量、作用機制進行研究,形成具有中醫藥特點的現代質量標準,已成為實現中藥標準化、現代化和國際化的一項重要課題。
目前,中藥的鑒別和質量控制的方法主要有:1)色譜法[1-2],研究和應用最廣泛的方法,氣相色譜法僅適用于分離、鑒別和分析其中揮發性成分,而高效液相色譜僅用于真溶液體系,需要復雜的前處理,使得藥物有效信息丟失,兩者均不能體現中藥的完整信息;2)波譜法[3-4],紫外吸收光譜主要用于中藥中所含的不飽和組分,紅外光譜與拉曼光譜也只能給出中藥的部分結構或所含官能團的信息,即圖譜難以全面反映中藥的特性和協同作用,核磁共振被認為是研究中藥有效成分化學結構的重要方法之一,但其制樣手續繁雜、測試成本高,同樣也僅為部分結構信息;3)DNA分子標記技術[5-6],該法可靠性強,重現性好,具有專屬性,但操作要求苛刻、儀器設備昂貴,還需引物篩選、標記、序列測定等,難以推廣普及。上述方法的應用范圍各有不可忽視的局限性,因此探索簡便、快速、高效的指紋圖譜新方法,以適應對中藥材的鑒別與質量控制,無疑是擺在分析工作者面前富有挑戰性的任務之一。
基于指紋圖譜技術建立的中藥質量控制體系應從其整體作用不同于化學藥的特點出發,實現對多組分構成的復雜體系的中藥全成分的有效分析,才能真正達到中藥的鑒別及質量控制,確保用藥安全、療效穩定[7]。
利用電化學方法與振蕩體系,獲取能顯示中藥特性的振蕩圖譜,并由圖譜反映的信息鑒別中藥種類和確定化學成分的量。方法靈敏、選擇性高、簡便可靠,已引起了高度重視[8-9]。本研究基于B-Z化學振蕩反應原理,構建了KBrO3-Ce(SO4)2-H2SO4-檸檬酸振蕩體系,通過對空白體系穩定性和重現性的考察與組成優化,用于27種中藥的電化學指紋圖譜的檢測,以及黃芪、枸杞子、白術道地性的鑒別,中藥形態和不同部位差異的區分等。結果表明,圖譜特異性顯著,信息豐富,可用于中藥的質量評價,道地性、真偽的鑒別及定量分析;同時也是對于色譜和波譜指紋圖譜技術的補充與拓展。
1 材料與方法
1.1 材料
CHI660b電化學工作站(上海辰華儀器公司),17型雙液接飽和甘汞電極、213型鉑電極(上海精密科學儀器有限公司),150 mL反應器(天津玻璃儀器廠)。
陳皮(四川)、五加皮(陜西)、西洋參(陜西)、川貝母(四川)、丹參(陜西)、川芎(四川)、杜仲(甘肅)、茯苓(河南)、葛根(廣西)、黃柏(四川)、肉桂(云南)、桑葉(陜西)、山楂(山東)等中藥材為市售,并經陜西省食品藥品檢驗研究院中藥室主任羅定強高級工程師鑒定為正品。硫酸高鈰、檸檬酸、氯化鉀、硝酸鉀、溴酸鉀、硫酸均為分析純,所有溶液用二次蒸餾水配制。
1.2 操作方法
以鉑電極為工作電極,雙液接飽和甘汞電極為參比電極。控制水浴溫度(37±1)℃,分別向反應器中加入1.00 g干燥的中藥粉末(粒徑<0.15 mm)、45 mL H2SO4溶液,攪拌下浸出15 min,再依次加入硫酸高鈰、檸檬酸,待基線穩定,最后加入5 mL KBrO3溶液,同時采集數據,記錄E-t振蕩曲線至電位振蕩消失。即獲得中藥非線性電化學指紋圖譜。
2 結果與討論
2.1 電化學指紋圖譜
根據化學振蕩原理,酸性條件下,BrO3-為氧化劑,檸檬酸為振蕩底物,硫酸高鈰為催化劑[10],而反應中生成具有自催化作用的HBrO2,其存在與否將直接導致BrO3-的還原產物Br-的形成和Ce4+→Ce3+的進行;當Br-累積與Ce4+消耗至臨界值時,將導致振蕩反應發生,表現為體系的氧化還原電位值隨時間呈周期性變化[11]。隨著有機底物檸檬酸的耗散,振蕩最終停止。電化學指紋圖譜中指紋峰的含義區別于色譜峰,色譜峰僅代表經色譜柱分離樣品后,單一組分隨保留時間的檢測結果,且不涉及任何化學反應,而振蕩波形的指紋峰則代表振蕩組分與底物反應隨時間的非線性變化,在振蕩圖譜中,1個指紋峰代表振蕩反應的1個周期。因此,電化學指紋圖譜可提供完整的定性及定量化學信息。
中藥為多組分復雜體系,其藥效為多種活性或非活性成分協同作用或“生克作用”。如以中藥為振蕩底物,固定振蕩體系各組分濃度,由于每種中藥參與振蕩反應的物質不同,對振蕩體系的擾動方式和程度亦不相同(表現為對振蕩反應的抑制或增強)。因此,利用這一規律可得到每種中藥的特征振蕩圖譜,從而實現對中藥的真偽鑒別及質量評價。
2.2 空白體系穩定性
中藥電化學指紋圖譜是其對空白振蕩體系的擾動而形成,疊加于空白振蕩圖譜之上。因此空白振蕩體系的穩定性是獲取準確結果的前提。本研究以誘導期(t0)、振蕩期(td)、最大振幅(Amax)、振蕩周期(T)、最高電位(Emax)、起始電位(Emin)、平衡電位(Ea)為考察指標,平行測定3次,其振蕩圖譜和參數分別見圖1和表1。表1中7組參數RSD≤1.63%,因此空白振蕩體系具有良好的穩定性和重現性。
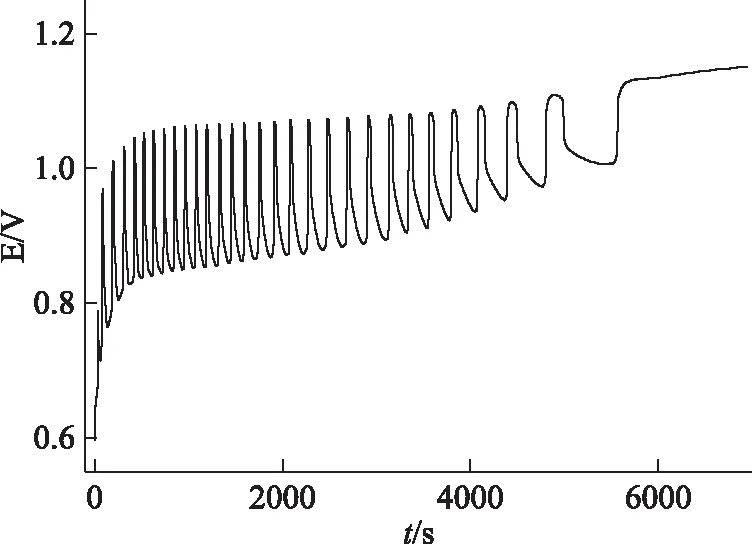
圖1 空白體系的振蕩圖譜
2.3 振蕩體系與測試條件
基于振蕩圖譜的7個特征參數,以振蕩波形特征顯著、信息完整、誘導期較短,振蕩期適中為依據,分別探討了體系各組分濃度、溫度、攪拌速率及中藥加入量對振蕩圖譜特征性的影響。經優化確定振蕩體系組成為:0.04 mol·L-1KBrO3、0.01 mol·L-1Ce(SO4)2、0.55 mol·L-1H2SO4、0.01 mol·L-1檸檬酸;檢測條件:溫度(37±1)℃,攪拌速率100 r·min-1,浸出時間15 min,中藥加入量1.0 g·(50 mL)-1。獲得穩定、重現的電化學指紋圖譜。
2.4 圖譜重現性
中藥指紋圖譜的特征之一,即重現性,也是其是否具有通用性和實用性的衡量標準。在確定的振蕩器及檢測條件下,獲取陳皮的電化學指紋圖譜,測定3次,考察其重現性,結果見表2。
如表2所示,陳皮電化學指紋圖譜7個參數的RSD≤2.61%,說明在相同測試條件下,同一中藥的振蕩圖譜具有較好的重現性。
2.5 圖譜特異性
在確定的振蕩體系和測試條件下,分別以黃芪、陳皮、白芍等27種中藥為振蕩底物,獲得形狀迥異的振蕩特征圖譜,見圖2。
圖2的振蕩圖譜波形各異,表現出因不同種類的中藥其化學成分不同而具有的個性特征,即特征參數存在顯著差異。因此,利用振蕩圖譜對中藥進行真偽辨識和質量評價是一種科學、有效的新方法。此法與色譜指紋圖譜相比,樣品無需預處理、操作簡單、檢測時間短,并且能夠給出中藥的群集信息,是一種有效的中藥鑒別手段和質量評價方法。
2.6 藥材道地性鑒別
道地藥材系指質優效佳,源于中醫藥臨床實踐,與自然條件和生態環境密切相關,是中藥學中控制藥材質量的一項獨具特色的綜合判別標準。目前,主要以經驗判斷或基源(DNA指紋圖譜)檢測等方法[7],前者缺乏科學性,而后者檢測手續繁雜、條件苛刻、成本高。為此,筆者以本研究所建立的振蕩體系對不同產地的白術、枸杞子和黃芪進行了檢測,獲得電化學指紋圖譜見圖3~4。
如圖3和圖4所示,不同源的同種中藥材的電化學指紋圖譜形狀相似,說明這些中藥對振蕩反應機理產生影響的化學成分相同,但Amax、Emin、T等有明顯差異。

表1 空白體系電化學指紋圖譜的穩定性
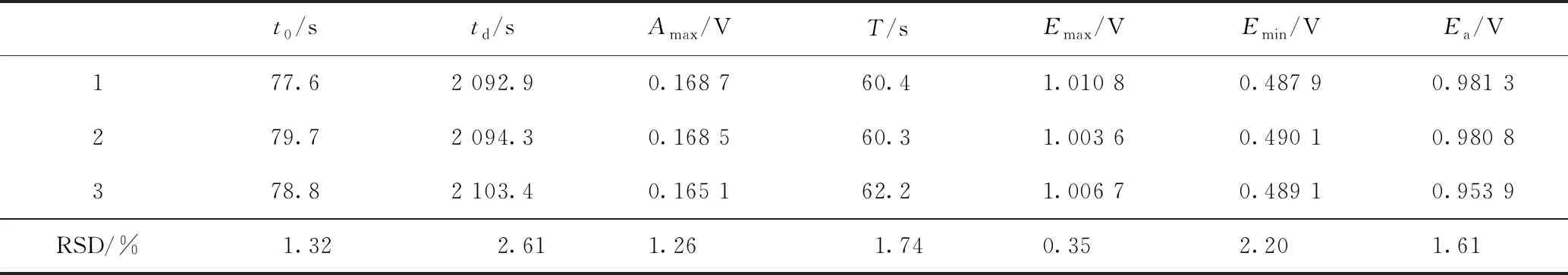
表2 陳皮電化學指紋圖譜的重現性

圖2 不同中藥的電化學指紋圖譜

圖3 不同產地中藥的電化學指紋圖譜

圖4 不同產地黃芪的電化學指紋圖譜
這些差異表明,盡管同種藥材所含活性成分的種類大致相同,但由于其生長的自然環境、耕種方式及采收季節等不同,導致不同地域的同種中藥活性成分含量存有差異,即定量信息之間存在明顯差異,從而對振蕩體系的擾動程度不同。因此,圖譜的特征參數存在差異。依此可見,電化學指紋圖譜用于中藥道地性的鑒別,科學、簡便、直觀。
2.7 中藥的質量評價
中藥振蕩圖譜的波形及內涵的特征參數,不僅與中藥所含化學成分的種類有關,還受到其含量多少的影響。在應用指紋圖譜技術對不同源的同種中藥進行質量評價時,要求其定性和定量信息之間均存在明顯差異。實驗測定了不同加入量黃芪的電化學指紋圖譜,在一定的檢測用量范圍內,考察定量信息與總活性成分含量的關系[12]。實驗發現,其td與m呈良好的線性關系(td=-2695m+7844,r=0.987),線性范圍1.0~1.5 g。
分別測試1.0 g內蒙古、甘肅、陜西黃芪的電化學指紋圖譜(見圖4),根據黃芪指紋圖譜td與m的關系,若以內蒙黃芪總活性成分為1,則陜西和甘肅黃芪的總活性成分分別為0.962 8、0.860 3。即總活性成分含量:內蒙古>陜西>甘肅,符合黃芪道地以“北芪最佳”的說法。在臨床應用中,3個產地的黃芪達到同一療效的用藥當量關系為:
內蒙古(1.000 g)=陜西(1.039 g)=甘肅(1.162 g)
2.8 不同部位中藥振蕩圖譜
中藥不同部位的化學成分及其含量存有差異,因此其各自的功效和藥用價值不同。如全當歸既能補血、又可活血,當歸尾則為破血,而當歸身偏補血和養血之功效。迄今,對同一中藥不同部位的振蕩圖譜鑒別尚未見到相關的報道。為此,分別測試了全當歸、當歸尾和當歸身的電化學指紋圖譜,見圖5。

圖5 當歸不同部位電化學指紋圖譜
由圖5可以看出,不同部位的當歸其指紋譜圖存在明顯差異,說明不同部位的當歸含有的有效成分和含量存在差別,對振蕩體系的擾動作用不同。因此,此法對同種中藥的不同部位可進行有效區分與評價,亦可為全藥材資源的綜合利用提供一定的科學依據。
2.9 中藥復方的指紋圖譜
中藥復方是指由2味或2味以上藥味的藥效相似或相近的中藥配伍,即所謂“相生”,是中醫方劑的主體組成部分。如茯苓的功效為利水滲濕、益脾和胃、寧心安神等,黃芪具有補氣生津、保肝、利尿等功效,故兩者可配伍使用;當歸與川芎均具有活血行氣、調經止痛的作用,可配伍使用。為此,檢測了2種復方中藥的電化學指紋圖譜(見圖6~7)。

圖6 黃芪+茯苓的電化學指紋圖譜

圖7 當歸+川芎的電化學指紋圖譜
圖6~7顯示,復方中藥的指紋圖譜與單味中藥的圖譜存在明顯差異,具有其特征性。這可能是由于復配后活性成分的協同作用對有機底物的擾動作用與單味中藥的活性成分存有差異,即對振蕩體系的擾動發生了改變。此為鑒別同藥效不同組方提供了一種可行的手段。
2.10 不同形態中藥指紋圖譜
中藥通常以水劑、散劑、丸劑等形態服用,實踐表明,中藥的形態與治療之間有著密切關聯。故選擇正確的形態服用,能充分發揮中藥的藥性及其臨床療效。為此,實驗對不同劑型陳皮的電化學指紋圖譜進行對比分析(見圖8)。

圖8 不同形態陳皮的電化學指紋圖譜
由圖8可以看出,同種藥材因所含活性成分相同而具有相似的指紋圖譜,但形態不同其圖譜間又存在明顯差異,對比空白振蕩體系可知,陳皮為抑制作用。圖8中陳皮汁的振幅Amax最小,抑制作用最強,振蕩期td長,藥效持續,即熬制使得中藥有效成分充分溶出。驗證了傳統服用中藥的正確性。
2.11 振蕩反應機理
振蕩反應通常分為能量積累、能量平衡、能量消耗3個階段,而溫度對各階段產生的影響不同。結果表明,反應速率隨著溫度的升高而加快,使得誘導期和振蕩期明顯縮短。由ln(1/t0)~T-1和ln(1/t)~T-1關系及Arrhenius式,可求得誘導期和振蕩期反應的活化能分別為58.2、67.7 J·mol-1,表明兩者反應機理不同。即振蕩反應包含誘導反應和振蕩反應2個歷程,誘導反應為振蕩反應積蓄能量,以使體系具有能夠發生振蕩反應的氧化還原電勢(φ氧化/φ還原>1.0 V,相較于飽和甘汞電極),兩者存在因果關系。
中藥的有效成分主要包含有多糖、皂苷、黃酮、生物堿、有機酸及微量元素等,化學成分十分復雜,少則數種,多者可達近百種。因此,本研究僅以參類藥材中常見的人參皂苷Rh2為例,推測其與振蕩體系的反應機理。
人參皂苷Rh2含有多個醇羥基、1個葡萄糖環和1個雙鍵,具有一定的還原性,且羥基可與酸發生酯化反應;在體系的催化作用下,可將醇羥基氧化成羰基[13]。根據B-Z振蕩理論推測機理如下:
過程1):Br-離子被氧化為HOBr


過程2):BrO3-被還原為HOBr2
過程3):Br-和Ce3+的再生

為了驗證振蕩機理和中藥對振蕩參數的擾動,利用Br-選擇性電極在線監測空白和加藥后振蕩器中c(Br-)的變化(見圖9~10)。圖9顯示,在空白振蕩體系中,c(Br-)的變化呈典型的B-Z化學振蕩特征,表明c(Br-)是振蕩反應發生的關鍵因素,控制著反應的進程和振蕩的循環過程。圖10表明,中藥的化學成分(人參皂苷Rh2)與Ce(SO4)2等發生反應,改變了振蕩組分的濃度,導致振蕩圖譜特征參數發生變化。在振蕩停止后空白體系c(Br-)逐漸升高,加藥體系則逐漸減小,推測是由于加入中藥發生化學反應所致。

圖9 空白振蕩體系的Br-振蕩圖譜

圖10 加藥振蕩體系的Br-振蕩圖譜
另外,采用循環伏安法測定了人參在該振蕩器中的循環伏安圖。結果表明,隨著人參加入量的增加,氧化峰電流值逐漸減小(見圖11),且與氧化峰電流呈良好的線性關系,r=0.991,說明人參中化學成分(人參皂苷Rh2)參與體系的振蕩反應,改變了振蕩器組分的濃度,由此也佐證了2.7所得td~m關系的正確性。

圖11 不同加藥量的循環伏安圖
3 結論
非線性電化學指紋圖譜既體現了中藥“君臣佐使、升降浮沉”的群集信息,又反映了不同中藥之間“相生相克”的協同效應,亦體現了以“整體性”和“模糊性”為顯著特證評價中藥材質量的要求。因此,可廣泛用于中藥的真偽識別、道地性鑒定以及質量的定性定量評價。
本研究建立KBrO3-Ce(SO4)2-H2SO4-檸檬酸振蕩體系及其最佳測試條件:0.04 mol·L-1KBrO3、0.01 mol·L-1Ce(SO4)2、0.55 mol·L-1H2SO4、0.01 mol·L-1檸檬酸,溫度(37±1)℃、攪拌速率100 r·min-1、浸出時間 15 min、中藥加入量1.0 g·(50 mL)-1。圖譜穩定重現、直觀明了、特異性顯著;方法簡便、快速、靈敏、有效,無需預處理試樣,具有推廣價值。
猜你喜歡
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國外匯(2019年17期)2019-11-16 09:31:14
基層中醫藥(2018年6期)2018-08-29 01:20:20
現代企業(2015年1期)2015-02-28 18:43:18
肝博士(2015年2期)2015-02-27 10:49:49
新高考·高一物理(2014年1期)2014-09-18 01:26:07
