兒科醫院首次發現新型遺傳性肝病,在疑難肝病領域開辟新研究方向
2020-09-03 13:53:48王炬亮復旦大學附屬兒科醫院編輯南溪
科學生活 2020年8期
關鍵詞:研究
文/王炬亮(復旦大學附屬兒科醫院) 編輯/南溪
隨著經濟社會的日益發展,兒童肝病的疾病譜在數十年間發生了巨大轉變,由原來以傳染性肝炎為主,逐漸轉變為以遺傳性肝病等罕見病為主。作為國家兒童醫學中心,復旦大學附屬兒科醫院的肝病專業團隊在多年的臨床工作中積累了豐富的經驗和研究成果。近日,兒科醫院的感染傳染科肝病研究團隊發現并報道了一種新的遺傳性肝病,致病基因為ZFYVE19,臨床表現為先天性肝纖維化、硬化性膽管炎和高GGT膽汁淤積癥。7月31日,該研究論文在線刊登于BMJ旗下的全球知名遺傳病雜志Journal of Medical Genetics,標志著復旦大學附屬兒科醫院肝病領域的專業研究水平登上了一個新的高峰。

圖1 論文ZFYVE19缺陷相關先天性肝纖維化、硬化性膽管炎和高GGT膽汁淤積癥由BMJ旗下的Journal of Medical Genetics在線發表。
據復旦大學附屬兒科醫院感染傳染科主任王建設教授介紹,感染傳染科肝病研究團隊近期發現并報道的新型遺傳性肝病,致病基因為ZFYVE19,臨床表現為先天性肝纖維化、硬化性膽管炎和高GGT膽汁淤積癥。這一成果由復旦大學附屬兒科醫院、復旦大學附屬金山醫院、中國科學院深圳先進技術研究院、復旦大學附屬華山醫院、上海交通大學附屬仁濟醫院、復旦大學生物醫學研究院及奧地利格拉茨大學醫學院病理系多家單位合作完成,并于7月31日在線刊登于BMJ旗下的全球知名遺傳病雜志Journal of Medical Genetics。該論文由課題組的欒維莎博士后、郝陳指博士和黎佳琪碩士為共同第一作者,王建設教授為通信作者。
王建設教授表示,探索真理的道路從來不是一帆風順的,創新性越強,研究難度越大,耗時越長。從初次通過二代測序方法從患者中篩選出ZFYVE19,到研究成果被認可并發表,歷時漫長的6年時間,經歷了多位研究團隊成員的接力奉獻。由于既往對于這一基因功能的研究十分有限,初期在國際學術會議進行報道時受到了業內學者的較多質疑。為了尋找致病證據,課題組研究人員積極收集更多病例、隨訪患者,提供具有說服力的家系證據;同時,與華山醫院、仁濟醫院合作,收集患者肝移植術中的組織樣本進行研究,證明在患者中該蛋白表達缺失(圖3);并與奧地利格拉茨大學醫學院病理系合作,分析研究患者肝組織的病理特征,從病理特征推測此病應該是纖毛病(圖4),終于找到進一步深入研究的線索;還與中科院深圳先進技術研究院合作,通過細胞系模型和患者來源的誘導干細胞實驗證實ZFYVE19變異影響纖毛形成,首次明確該基因與纖毛形成的直接相關性(圖5);最終,在與復旦大學生物醫學研究院合作中,發現ZFYVE19基因的數據庫注釋存在缺陷,從而導致這一基因在測序分析中容易被忽略,最后我們通過該基因轉錄和蛋白翻譯方面進行深入研究,對該基因的翻譯起始位點進行了新的注釋,克服了注釋錯誤對基因功能預測造成的誤導誤判(圖6),故而至今才被學術界揭示。在多方協助、共同努力下,課題組克服重重困難,歷時多年的研究成果終得以獲得業內專家認可。
豐碩的成果總來源于長期的努力,作為國內最大的兒童肝病診治和研究中心,王建設教授帶領肝病臨床及科研團隊,從2003年開始就專注于兒童膽汁淤積癥的臨床和研究,且在國際上較早在患者中應用二代測序方法進行已知疾病的診斷和未知疾病的發掘,在多年臨床和科研工作中積累了豐富的臨床病例和研究數據資源。2017年王建設教授領銜的肝病團隊鑒定了MYO5B缺陷引起的低GGT膽汁淤積癥譜系,被國際上公認為家族性膽汁淤積癥6型(圖7)。今年3月,該團隊又在國際上率先報道了一組由USP53缺陷引起低GGT膽汁淤積癥譜系,詳細闡述了該病的臨床特征及病理特征,并揭示了這一新基因與已知低GGT膽汁淤積癥致病基因TJP2的潛在關聯(圖8)。而近期報道的ZFYVE19則是全球首次關于該基因致病的報道,臨床上表現為先天性肝纖維化、硬化性膽管炎和高GGT膽汁淤積癥。在未來,基于國家兒童醫學中心的大平臺,相信復旦大學附屬兒科醫院兒童肝病中心這一研究成果必將為治療疑難肝病領域開辟新的道路,服務于更多疑難肝病患兒,為更多無助的家庭帶來希望和慰藉。
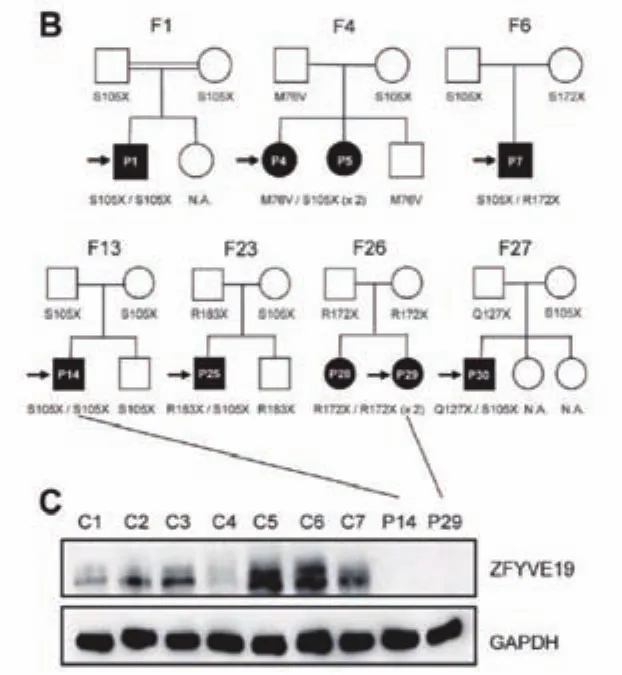
圖3 ZFYVE19缺陷患者家系圖顯示符合常染色體隱性遺傳,患者肝組織中ZFYVE19蛋白缺失。
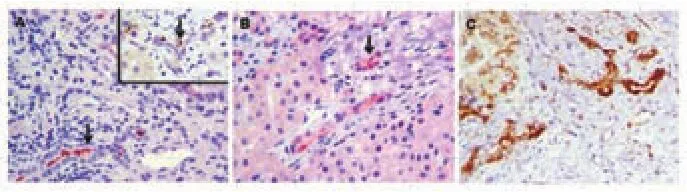
圖4 患者肝臟組織病理中纖毛蛋白表達異常,提示纖毛病可能。
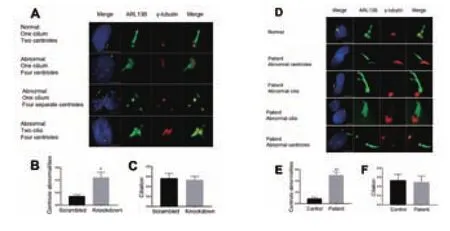
圖5 細胞實驗證實,敲低ZFYVE19可以使細胞形成異常數目和形態的中心粒及纖毛。
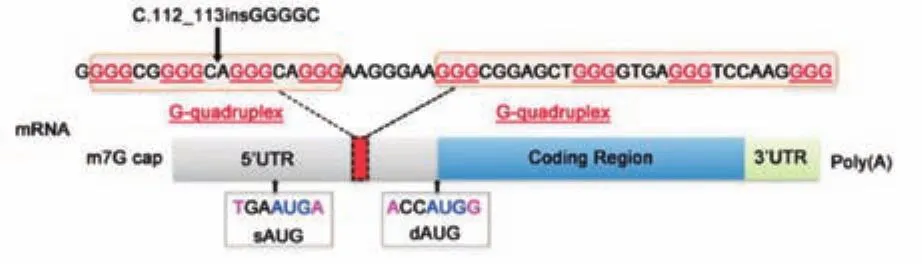
圖6 ZFYVE19的mRNA結構重新分析發現以往數據庫中注釋蛋白翻譯起始位點錯誤。sAUG:注釋翻譯起始位點。dAUG:實際翻譯起始位點。

圖7 MYO5B缺陷導致低GGT膽汁淤積癥疾病譜系,發表于2017年Hepatology。

圖8 USP53缺陷導致低GGT肝內膽汁淤積癥的臨床特點、病理特征及超微結構,2020年發表于Liver International。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
