多指標正交試驗優選葆腎合劑最佳提取工藝*
2020-09-10 03:27:24馮子芳袁曉航陳曉偉費倩倩費逸明
中國藥業 2020年17期
馮子芳,袁曉航,陳曉偉,費倩倩,費逸明
(南京中醫藥大學附屬無錫市中醫醫院,江蘇 無錫 214000)
葆腎合劑是治療早中期慢性腎臟病的經驗方,由芡實、黃芪、石韋、金櫻子、覆盆子等組方,補腎兼顧健脾,以補后天而實先天,固攝之時不忘清利,攝而不滯,利而不傷。中藥制劑通過多成分、多靶點協同發揮整體藥效,因此,多指標綜合評分是優化提取工藝的重要方法[1-2]。芡實益腎固攝[3-4],黃芪益氣固攝,米仁根清利濕熱,但相關研究少,故不選擇其作為含量測定的成分。黃芪中毛蕊異黃酮葡萄糖苷為其有效成分,相關檢測手段成熟,分離度好[5-7]。覆盆子中的鞣花酸、石韋中的綠原酸均為藥材主要有效成分,理化性質穩定。故選擇毛蕊異黃酮葡萄糖苷、鞣花酸、綠原酸作為含量測定指標成分。本試驗中以有效成分含量和浸膏得率為考察指標,采用多指標綜合評分方法[8]優選最佳提取工藝,明確葆腎合劑的物質基礎。現報道如下。
1 儀器與試藥
1.1 儀器
Agilent 1200 型高效液相色譜儀,包括DAD 二極管陣列檢測器(美國Agilent 公司);BSA224S-CW 型電子天平(德國Sartorius 公司,精度為十萬分之一);KQ-100 型超聲波清洗機(昆山市超聲儀器有限公司,功率為100 W,頻率為40 kHz)。
1.2 試藥
毛蕊異黃酮葡萄糖苷對照品(批號為111920-201606),鞣花酸對照品(批號為111959-201602),綠原酸對照品(批號為110753-201817),均購自中國食品藥品檢定研究院;中藥飲片(產品信息見表1)經無錫市中醫醫院副主任中藥師胡敏敏鑒定為合格品;葆腎合劑(醫院制劑,批號分別為191201,191202,191203);乙腈為色譜純,水為超純水,其余試劑均為分析純。

表1 中藥飲片產品信息
2 方法與結果
2.1 含量測定
2.1.1 色譜條件與系統適用性試驗
色譜柱:WatersXbridgeC18柱(250mm×4.6mm,5μm);柱溫:30 ℃;流動相:乙腈(A)-0.2%磷酸溶液(B),梯度洗脫,0~10 min 時92%B,10~12 min 時92%B→89%B,12~50 min 時89%B;檢測波長:326 nm(綠原酸),254 nm(鞣花酸和毛蕊異黃酮葡萄糖苷),采用雙波長檢測;流速:1.0 mL/min;進樣量:10 μL。在此色譜條件下,毛蕊異黃酮葡萄糖苷峰、鞣花酸峰和綠原酸峰與其他峰達基線分離。色譜圖見圖1。

圖1 高效液相色譜圖
2.1.2 溶液制備
取毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸對照品適量,精密稱定,加甲醇溶解,配制成含毛蕊異黃酮葡萄糖苷32 μg/mL、鞣花酸157.8 μg/mL、綠原酸53 μg/mL的混合對照品溶液。精密量取3 批樣品(批號分別為191201,191202,191203)1.5 mL,置5 mL 容量瓶中,加甲醇稀釋至刻度,超聲(功率為100 W,頻率為40 kHz)10 min,放冷,加甲醇定容,搖勻,用0.22 μm 微孔濾膜濾過,取續濾液,即得供試品溶液。按葆腎合劑制備工藝,分別制備缺黃芪、缺覆盆子、缺石韋的陰性樣品,按供試品溶液制備方法制備陰性對照品溶液。
2.1.3 方法學考察
線性關系考察:分別精密移取上述混合對照品溶液適量,加甲醇溶解,稀釋成不同質量濃度的混合對照品溶液,0.22 μm 微孔濾膜濾過,分別精密吸取10 μL,注入液相色譜儀,按擬訂色譜條件進樣測定,記錄峰面積,以各對照品質量濃度(X)為橫坐標、峰面積(Y)為縱坐標繪制標準曲線。結果見表2。
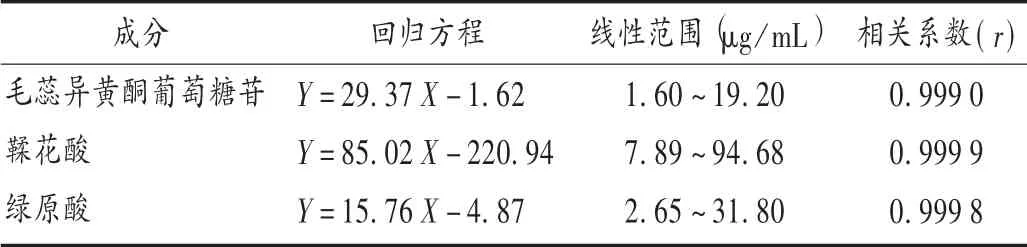
表2 線性關系考察結果
精密度試驗:取同一對照品溶液適量,連續進樣6 次,記錄毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸的峰面積。結果的RSD 分別為0.44%,0.27%,0.33%(n=6),表明儀器精密度良好。
重復性試驗:取樣品(批號為191202)適量,依法平行制備6 份供試品溶液。精密吸取10 μL 分別進樣測定。結果毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸峰面積的RSD 分別為1.72%,2.09%,1.42%(n=6),表明方法重復性良好。
穩定性試驗:取重復性試驗項下同一供試品溶液,分別于0,1,2,4,8,16 h 時按擬訂色譜條件進樣測定,每次10 μL。結果毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸峰面積的RSD 分別為0.79%,1.98%,2.63%(n=6),表明供試品溶液在16 h 內穩定。
加樣回收試驗:精密量取樣品(批號為191202)0.75 mL,按1 ∶1(供試品溶液和混合對照品溶液中有效成分質量比)精密加入混合對照品溶液0.5 mL,依法平行制備6 份供試品溶液,按擬訂色譜條件進樣測定,記錄峰面積。結果毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸的平均加樣回收率分別為98.15%,97.08%,97.82%,RSD 分別為2.51%,2.19%,2.52%(n=6)。
2.2 正交試驗設計
試驗方案:按處方量稱取全方藥材,共72.5 g,分別取9 份。以毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸的量為有效成分指標,采用正交設計試驗L9(34),考察煎煮時間(因素A)、煎煮次數(因素B)、加水量(因素C)和浸泡時間(因素D)對提取效果的影響,因素水平見表3。按正交試驗方案,過濾、合并提取液,濾液濃縮至200 mL,得9 個正交試驗的樣品溶液。
浸膏得率測定:精密量取樣品溶液25 mL,置干燥至恒定質量的蒸發皿中,水浴蒸干,于105 ℃烘箱中干燥至恒重。結果見表4。

表3 正交試驗因素水平表
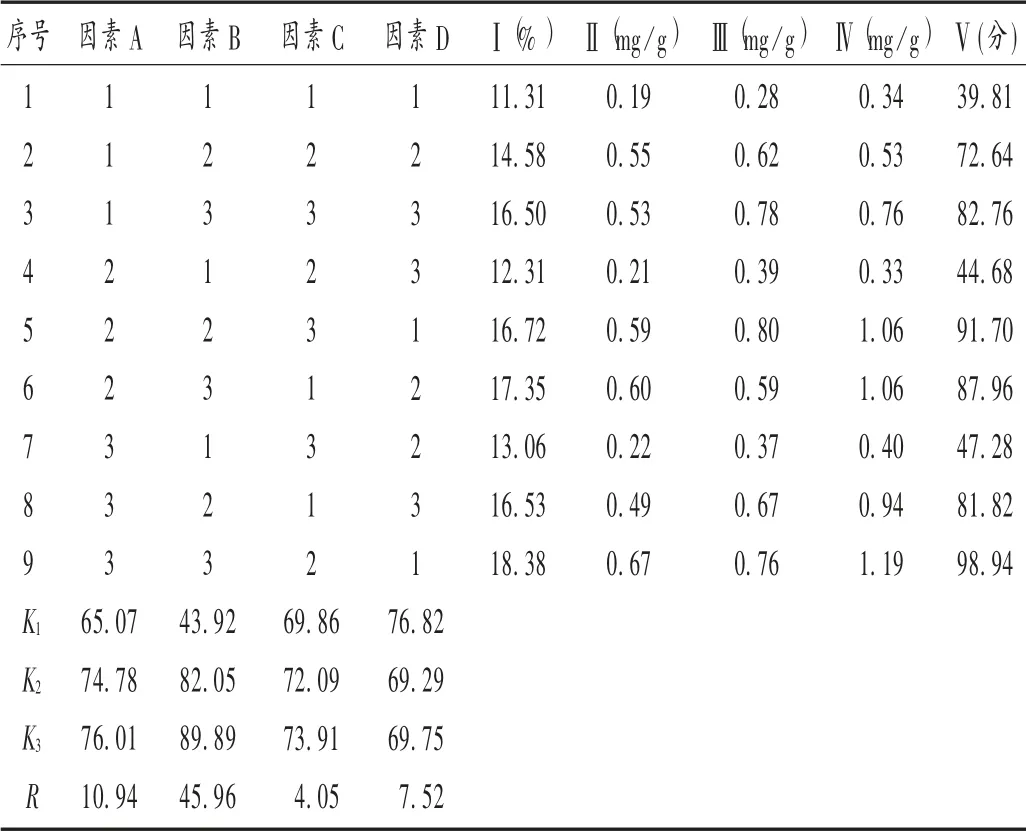
表4 水提工藝正交試驗結果
有效成分含量測定:依法制備供試品溶液,按擬訂色譜條件,分別測定9 份正交試驗供試品溶液中毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸含量,各成分的質量分數以各單味藥材投料量計,如鞣花酸質量分數=鞣花酸提出量(mg)/覆盆子投料量(g)。結果見表4。
綜合評價及結果分析:以有效成分的質量分數和浸膏得率進行綜合評分,評分時以各指標的試驗最大值為參照,將數據進行歸一化,再給以不同權重[9],參考君臣佐使分配權重,綜合評分=(樣品浸膏得率/樣品中最大浸膏得率)×30% ×100+(樣品毛蕊異黃酮葡萄糖苷質量分數/樣品中最高毛蕊異黃酮葡萄糖苷質量分數)×30% ×100+(樣品鞣花酸質量分數/樣品中最高鞣花酸質量分數)×20% ×100+(樣品綠原酸質量分數/樣品中最高綠原酸質量分數)×20% ×100,綜合評分越高,表明提取效果越好。以綜合評分對試驗結果進行直觀分析和方差分析[10],結果見表4 和表5。由表4 可見,各因素影響順序為煎煮次數>煎煮時間>浸泡時間>加水量;A3>A2>A1,B3>B2>B1,C3>C2>C1,D1>D3>D2,優選水平組合為A3B3C3D1。由表5 可見,煎煮次數對提取影響顯著(P<0.05),而加水量、煎煮時間和浸泡時間均無顯著影響。故確定最佳工藝為加10 倍量水,浸泡30 min,提取3 次,每次2.0 h。

表5 方差分析結果
2.3 驗證試驗
為驗證工藝的合理性和穩定性,需進行驗證試驗[11]。按處方比例稱取3 份藥材72.5 g,加10 倍量水浸泡30 min,回流提取3 次,每次2.0 h,合并濾液,濃縮至200 mL。測得浸膏得率和毛蕊異黃酮葡萄糖苷、鞣花酸、綠原酸含量見表6。毛蕊異黃酮葡萄糖苷、鞣花酸和綠原酸含量的RSD 分別為1.66%,0.79%,2.10%(n=3),表明工藝穩定可行、重復性好。
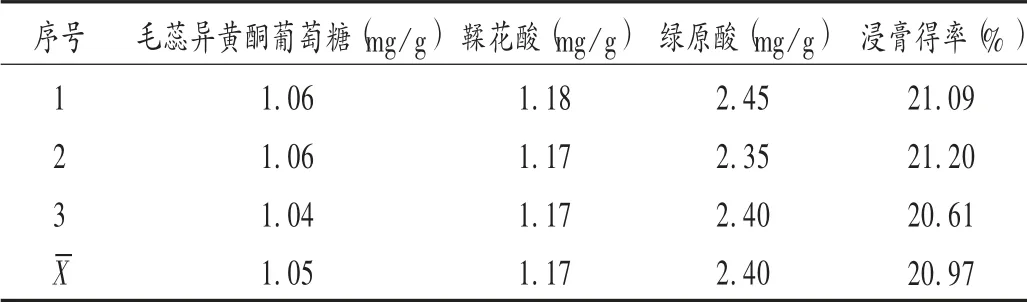
表6 最佳工藝驗證試驗結果(n=3)
3 討論
多指標綜合評價是優化中藥復方提取工藝的重要手段,設計正交試驗優化提取工藝時,根據影響因素對提取工藝的貢獻大小分配不同的權重系數[12-13]。黃芪為方中臣藥,而毛蕊異黃酮葡萄糖苷為《中國藥典》規定的含量測定成分,給予其30%的權重;覆盆子中鞣花酸類化合物具有抗氧化、抗菌等作用[14],給予20%的權重;石韋中綠原酸有抗菌、降壓等作用[15],給予20%的權重;同時,浸膏得率也是提取工藝考察的重要指標,故將其權重設為30%。這既保證了數據指標的全面性,又根據各指標的影響程度給予了不同的側重,保證分析結果合理、科學[16]。
通過查閱文獻及多次預試驗摸索,本試驗中采用雙波長切換的方法,實現了同時測定葆腎合劑中3 種有效成分。通過對比不同流動相系統對分離效果的影響,選擇了乙腈-水、乙腈-0.2%磷酸溶液、乙腈-0.2%甲酸溶液等流動相。結果表明,選擇乙腈-0.2%磷酸溶液為流動相,梯度洗脫時,所測成分各峰與其他峰有良好的分離度。利用3 種不同品牌的色譜柱Waters XBridge C18柱(250 mm×4.6 mm,5 μm),YMC-Pack ODS-A C18柱(250 mm×4.6 mm,5 μm),Agilent Eclipse XDB C18柱(250 mm×4.6 mm,5 μm)對3 種成分進行了含量測定,分離度較好。經方法學考察證實,該方法可達到有效成分分離測定的要求,且準確、可靠。
在葆腎合劑提取工藝的試驗研究中,充分考慮了君臣佐使的配伍原則、主要成分的化學性質及規模化生產的要求,采用多指標綜合評分法,優選最佳提取工藝,并進行了驗證,避免了單一因素對中藥復方提取影響的局限性和片面性。
