PPARγ激動劑對COPD大鼠炎癥及血管重塑指標的影響
2020-09-11 07:35:44王琛,李智,譚林
中國臨床醫學 2020年4期
王 琛, 李 智, 譚 林
青島大學附屬青島市市立醫院重癥醫學科,青島 266071
根據最新的《全球疾病負擔》報告,目前所有非傳染性慢性肺部疾病加在一起構成全球非傳染性疾病的第3大死因,其中2016年慢性阻塞性肺疾病(COPD)造成230萬人死亡[1]。在COPD中,肺動脈高壓(pulmonary hypertension, PH)的患病率與病情的嚴重程度密切相關,多達90%的患者靜息平均肺動脈壓(mPAP)超過20 mmHg(1 mmHg=0.133 kPa)。約有1%的COPD患者患有嚴重的PH,mPAP值在35~40 mmHg,PH在COPD中的存在和嚴重程度對患者生存率有重要影響。缺氧引發的肺血管收縮是PH的主要致病機制,持續的血管收縮導致肺血管結構的變化,主要影響肺小動脈。在COPD中,炎癥、低氧、煙草煙霧的暴露導致血管重塑、肺血流動力學改變[2]。
過氧化物酶增殖物活化受體γ(peroxisome proliferator activated receptorγ, PPARγ)的激動劑羅格列酮在COPD患者中給藥時可抑制香煙煙霧誘導的支氣管上皮細胞炎癥因子、趨化因子和活性氧類(reactive oxygen species,ROS)等的產生,且羅格列酮可劑量依賴性地抑制脂多糖(lipopolysaccharide,LPS)誘導的腫瘤壞死因子(tumor necrosis factor-α,TNF-α)產生,PPARγ參與COPD的氣道炎癥過程[3],但針對其在COPD患者炎癥因子白介素-8(IL-8)的產生和肺動脈的血管重塑的作用研究尚少。肺小動脈血管的重塑是導致PH的主要原因,目前COPD合并PH患者的治療仍以針對誘因治療為主,臨床治療效果不佳,個體化差異較大。為此,本研究擬探討鹽酸羅格列酮調控PPARγ轉錄水平對COPD大鼠炎癥因子的產生和肺小動脈血管重塑的影響。
1 材料與方法
1.1 主要儀器及試劑 雄性Wistar大鼠36只,購自青島市藥物檢驗檢疫所,6~8周齡,體質量180~220 g,SPF級飼養。試劑:哈德門(山東中煙工業有限責任公司,中國);鹽酸羅格列酮(文迪雅,4 mg/片);LPS(美國Sigma公司);大鼠白介素-8(IL-8) ELISA檢測試劑盒(南京建成生物工程研究所,中國);TRIzol試劑、M-MuLV一步法RT-PCR試劑盒、引物(上海生工生物工程技術服務有限公司)。主要儀器:自制煙熏箱和低氧倉。低氧倉大小約54 cm×30 cm×32 cm,鉆5個直徑3 cm孔,使倉內保持常壓,并往倉內注入氮氣調節氧濃度,使氧體積分數維持在0.1左右。
1.2 動物分組及處理 將大鼠按數字法隨機分為藥物治療組(A組)、非藥物治療組(B組)和空白對照組(C組),每組12只。A組參照鐘小寧等[4]方法制作COPD肺動脈高壓大鼠模型,B組與A組相似,但第3周開始每只大鼠每天給予鹽酸羅格列酮1 mg治療;C組給予相同體積生理鹽水模擬造模,正常飼養。
1.3 組織學檢查 以100 g/L水合氯醛(4 mL/kg)腹腔注射麻醉大鼠后,打開胸腔,取雙肺分別置于-70℃冰箱和40 g/L中性甲醛中保存備用。肺組織以40 g/L中性甲醛固定后,常規脫水包埋,并進行蘇木精-伊紅染色。每只大鼠肺組織切片隨機選擇5個視野,按照文獻[5]的方法觀測直徑50~100 μm的腺泡內肌化型小動脈的血管壁面積、血管總面積,并計算血管壁面積/血管總面積(MA%)比值。
1.4 IL-8檢測 準備好標準品并用洗滌緩沖液1∶1稀釋于反應孔內,后取大鼠肺組織50 mg,加入適量生理鹽水搗碎,離心10 min后取待測樣品上清液50 μL置于反應孔內,立即加入50 μL生物素標記的抗體,蓋上膜板,輕輕振蕩混勻,37℃溫育1 h,甩去孔內液體,每孔加滿洗滌液,震蕩30 s,甩去洗滌液,用吸水紙拍干,重復此操作3次后,每孔加入80 μL的親和鏈酶素-HRP,輕輕振蕩混勻,37℃溫育30 min;甩去孔內液體,每孔加滿洗滌液,振蕩30 s,甩去洗滌液,用吸水紙拍干,重復此操作3次后,每孔加入底物A、B各50 μL,輕輕振蕩混勻,37℃溫育10 min,避免光照,取出酶標板,迅速加入50 μL終止液,加入終止液后立即在450 nm波長處測定各孔的光密度(D)值,以D值為縱坐標,相應的待測物質濃度為橫坐標,繪制相應曲線,并根據其D值由標準曲線換算出相應濃度。
1.5 RT-PCR檢測PPAR mRNA的轉錄水平 取肺組織50 mg,加入0.5 mL TRIzol提取總RNA,測定RNA濃度。取1 μL mRNA產物按照一步法反轉錄試劑盒說明書合成cDNA,以此為模板行PCR擴增。引物通過Primer Premier5軟件設計,由上海生工生物工程有限公司合成。PPAR上游引物序列為5′-T CTCT CCGTAAT GGAAGACC-3′,下游引物序列為5′-GCAT TAT GA GACAT CCCCAC-3′;actin上游引物序列為5′-AGCCAT GTAGCCA TCC-3′,下游引物序列為5′-AGCCAT GTAGCCA TCC-3′。將PT-PCR產物在12 g/L的瓊脂糖凝膠中電泳(100 V)30 min,在紫外燈下檢測目的條帶電泳情況,應用Scion image軟件對電泳條帶進行吸光度掃描,以面積吸光度表示條帶的豐度,以此作為mRNA表達的強度,以actin作為內參照,PPARγ mRNA的相對表達水平采用PPARγ/actin計算得出。
1.6 統計學處理 采用SPSS 17.0進行統計處理。組間死亡率比較采用Fisher確切概率法,PPARγ轉錄水平、MA%與IL-8值多組間比較采用單因素方差分析LSD法,組間相關性分析采用Pearson相關性分析,檢驗水準(α)為0.05。
2 結 果
2.1 存活率分析 A組有2只大鼠分別在第5、6周死亡;B組、C組大鼠無死亡。因樣本量<40,所以采用Fisher確切概率法進行計算。A組大鼠存活率低于C組,組間差異無統計學意義(Fisher值=3.856,P=0.167)。
2.2 血管壁病理改變 病理切片結果(圖1)示:A組大鼠血管壁周圍中性粒細胞增多,即淋巴細胞形成,管壁平滑肌層增厚,其外觀可見大量膠原沉積。C組大鼠血管壁周圍無明顯炎性細胞浸潤,血管壁平滑肌層未見增厚,無膠原沉積。B組血管壁重塑及炎性浸潤情況較A組減輕,但仍較C組嚴重。病理切片由經驗豐富的病理科醫師雙盲讀片。光鏡下病理評分基于以下變量:肺小動脈周圍中性粒細胞浸潤、血管平滑肌層增厚程度。嚴重程度評分:無損傷=1分(無病理改變);輕度損傷=2分(有中性粒細胞浸潤,且血管壁輕度增厚);重度損傷=3分(大量中性粒細胞浸潤且血管壁明顯增厚)。每個樣本有5個切片,被隨機分析,以平均值作為樣本的最終評分。結果見圖1。

圖1 大鼠肺組織病理切片評分
2.3 肺小動脈血管重塑比較 對直徑50~100 μm肌化性動脈進行分析,結果(表1)顯示:A組、B組、C組間MA%差異具有統計學意義(F=47.84,P<0.01),A組MA%較C組明顯升高,B組MA%較A組明顯下降(P均<0.05)。
2.4 IL-8檢測指標比較 結果(表1)表明:A組、B組、C組之間IL-8差異具有統計學意義(F=47.87,P<0.01),A組明顯高于B組,B組高于C組(P均<0.05)。
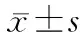
表1 A、B、C組MA%和IL-8比較
2.5 肺組織PPARγ轉錄水平比較 結果(圖2)表明:A、B、C組之間差異具有統計學意義(F=96.39,P<0.01),C組、B組、A組PPARγ/actin指標逐漸下降,A組明顯高于B組,B組高于C組(P均<0.05)。

圖2 3組PPARγ/actin水平比較
2.6 PPARγ轉錄水平與MA%、IL-8值的相關性 B組大鼠中PPARγ轉錄水平與MA%負相關(r=-0.662,P<0.05),與IL-8值負相關(r=-0.615,P<0.05),而B組中MA%與IL-8正相關(r=0.980,P<0.05)。
3 討 論
COPD是感染引起的、進行性氣道炎癥,慢性氣道炎癥在COPD中起主要作用,可引起杯狀細胞增加、黏液腺增生、肉芽腫和肺氣腫,導致巨噬細胞激活并釋放TNF-α、IL-6、IL-8、單核細胞趨化肽-1(MCP-1)、白三烯B-4和ROS等炎癥介質和趨化因子。目前氣道上皮細胞已被證實可分泌IL-8,引起小氣道纖維化進展,是COPD的炎性標志物。另有國外研究[6]表明,IL-8是使中性粒細胞聚集的主要細胞因子,其也被認為是COPD發展的關鍵因素。香煙煙霧足以刺激COPD氣道中IL-8的過度釋放,且在COPD急性加重期時,中性粒細胞和循環中的IL-8可在氣道中大量聚集。
在COPD慢性炎癥的損傷與修復過程中,肺小動脈血管平滑肌的增殖和血管壁周圍大量的膠原沉積共同參與了肺血管壁結構的重建[7-8]。目前作為PPARγ激動劑的噻唑烷二酮(TZD),其與糖皮質激素受體相似,是具有抗氧化作用的核激素受體超家族成員之一,通過多種機制發揮有效的抗氧化和抗炎作用,包括下調NF-κB和其他促炎轉錄因子[3],雖然其主要作為新型胰島素增敏劑,被廣泛用于2型糖尿病的治療,但也在多種肺細胞類型中普遍表達,包括肺血管內皮細胞和平滑肌細胞。在實驗性肺高壓動物模型(主要通過低氧誘導肺高壓)中,肺和肺小動脈中的PPARγ表達下調,導致發展為PH,這提示PPARγ在PH中具有保護作用。PPARγ激動劑TZD藥物羅格列酮激活PPARγ是對抗PH的有前途治療劑,可以保護肺小動脈,部分原因是可以通過抑制肺動脈平滑肌細胞增殖和遷移而實現該作用[9]。
本研究主要探討PPARγ激動劑是否在低氧、煙熏、LPS誘導下的COPD大鼠肺組織中有抑制促炎因子IL-8的生成和改善血管重塑的作用。病理切片顯示,B組肺血管壁重塑及其周圍的中性粒細胞浸潤較A組減輕,但仍較C組嚴重;相關實驗結果顯示,A組、B組大鼠的MA%比值、IL-8均高于C組,A組大鼠明顯高于B組。A、B、C組肺組織內PPARγ的轉錄水平依次遞增,B組大鼠PPARγ轉錄水平與MA%比值、IL-8均負相關。以上結果說明PPARγ激動劑羅格列酮可以抑制肺血管平滑肌細胞的增殖、抑制血管結構重塑、抑制促炎因子IL-8生成,抑制IL-8的生成可能抑制了肺血管周圍中性粒細胞的浸潤,而肺部炎癥是COPD進展的核心機制[10],那么本研究表明,羅格列酮可能是通過抑制其血管周圍的炎癥、中性粒細胞的浸潤,從而抑制血管壁的重塑和膠原蛋白的沉積,但是其中的具體生物學機制如何尚不清楚,這為進一步的研究提供了方向。
綜上所述,羅格列酮在COPD大鼠中可通過激活PPARγ而有效抑制IL-8的生成,從而抑制肺血管壁平滑肌細胞的增殖和膠原蛋白的沉積,這為羅格列酮治療COPD肺高壓提供了一定的理論支持,為臨床探索治療肺高壓的新型藥物提供了新思路。
