尪痹片HPLC 指紋圖譜建立
2020-09-15 03:17:52於慧佳董紅陽孫立新
中成藥 2020年8期
關鍵詞:質量
於慧佳 董紅陽 孫立新
(沈陽藥科大學藥學院, 遼寧本溪117004)
尪(音同“王” ) 痹片由地黃、熟地黃、續斷、附子(黑順片)、獨活、骨碎補、桂枝、淫羊藿、防風、威靈仙、皂角刺、羊骨、白芍、狗脊 (制)、知母、伸筋草、紅花17 味藥材組成,方中續斷、骨碎補、狗脊、羊骨、附子、淫羊藿、地黃、熟地黃共為君藥,桂枝、獨活、防風、威靈仙共為臣藥,白芍、知母、伸筋草、皂角刺、紅花共為佐藥,可治療肝腎不足、風濕阻絡所致的尪痹,癥見肌肉、關節疼痛等,以及類風濕性關節炎見有上述證候者[1],它具有鎮痛抗炎等藥理作用[2?6],對多種風濕骨病行之有效[7?15]。在2015 年版《中國藥典》 一部中,尪痹片質量控制僅規定了TLC 定性鑒別和淫羊藿苷含有量測定,無法全面控制該制劑質量。
中藥指紋圖譜能反映中藥可測成分的整體質量信息,整體評價產品質量的穩定性和一致性[16]。因此,本實驗建立尪痹片HPLC 指紋圖譜,并通過主成分分析評價該制劑質量。
1 材料
1.1 儀器 LC?20AT 型高效液相色譜儀(配置SPD?M20A型二極管陣列檢測器、CBM?20A 型控制器、Labsolution 色譜工作站,日本島津公司);JF1004 型電子天平(萬分之一,余姚市金諾天平儀器有限公司);TG332A 型微量分析天平(十萬分之一,湘儀天平儀器廠);KQ5200B 型超聲波清洗器(昆山超聲儀器有限公司)。
1.2 試劑與藥物 尪痹片[遼寧好護士藥業(集團) 有限公司,批號161007、161116、160710、161018、161216、170209、161209、160315、151208、160605,編號S1~S10,0.5 g/片]。原兒茶酸(批號110809?201205)、原兒茶醛(批號110810?201608)、芍藥苷(批號110736?201741)、淫羊藿苷(批號110737?201516) 對照品均購自中國食品藥品檢定研究院;馬錢苷酸(批號M?008?180102) 對照品購自成都瑞芬思生物科技有限公司。乙酸、甲醇、乙腈為色譜純(百靈威科技有限公司);純化水(杭州娃哈哈集團有限公司)。
2 方法與結果
2.1 色譜條件與系統適用性試驗 Diamonsil C18色譜柱(250 mm×4.6 mm,5 μm);流動相乙腈(A) -0.1%乙酸(B),梯度洗脫(0~12 min,5%~10% A;12~16 min,10%A;16~18 min,10%~12%A;18~33 min,12%~14%A;33~40 min,14%~18% A;40~60 min,18%~25% A;60~80 min,25%~40%A;80~90 min,40%~80%A;90~95 min,80%A);體積流量1.0 mL/min;柱溫30 ℃;檢測波長235 nm;進樣量5 μL。在此條件下,理論塔板數按各成分計均不低于5 000,各成分色譜峰與相鄰峰的分離度均大于1.2。
2.2 供試品溶液制備 將片劑去包衣后研細,精密稱取粉末1.0 g,置于具塞錐形瓶中,精密加入20 mL 25%甲醇,密塞,稱定質量,超聲(250 W、33 kHz) 提取30 min,放冷,25%甲醇補足減失的質量,搖勻,0.45 μm 微孔濾膜過濾,取續濾液,即得。
2.3 對照品溶液制備 精密稱取各對照品適量,25%甲醇制成分別含原兒茶酸103.5 μg/mL、馬錢苷酸398.9 μg/mL、原兒茶醛47.9 μg/mL、芍藥苷1 910.2 μg/mL、淫羊藿苷179.0 μg/mL 的溶液,即得。
2.4 方法學考察
2.4.1 精密度試驗 取同一份供試品溶液 (批號161007),在“2.1” 項色譜條件下進樣測定6 次,以6 號峰(馬錢苷酸) 為參照測定各共有峰相對保留時間、相對峰面積RSD,測得兩者分別為0.16%~1.2%、0.83%~1.9%,表明儀器精密度良好。
2.4.2 重復性試驗 取同一批樣品 (批號161007) 6份,按“2.2” 項下方法制備供試品溶液,在“2.1” 項色譜條件下進樣測定,以6 號峰(馬錢苷酸) 為參照測定各共有峰相對保留時間、相對峰面積RSD,測得兩者分別為0.14%~0.83%、0.97%~1.9%,表明該方法重復性良好。
2.4.3 穩定性試驗 取同一份供試品溶液 (批號161007),于0、4、8、12、24 h 在“2.1” 項色譜條件下進樣測定,以6 號峰(馬錢苷酸) 為參照測定各共有峰相對保留時間、相對峰面積RSD,測得兩者分別為0.13%~1.5%、0.69%~1.8%,表明供試品溶液在24 h 內穩定性良好。
2.5 指紋圖譜生成 取10 批片劑,按“2.2” 項下方法制備供試品溶液,在“2.1” 項色譜條件下進樣測定,建立指紋圖譜,結果見圖1,再采用中藥指紋圖譜相似度評價系統(2012 版) 生成對照指紋圖譜,發現有26 個共有峰,見圖2,其中6 號峰(馬錢苷酸) 分離度較好,峰面積較穩定,故選擇其作為參照峰(S)。經與對照品比對后,峰5、6、7、14、22 分別為原兒茶酸、馬錢苷酸、原兒茶醛、芍藥苷、淫羊藿苷。

圖1 10 批樣品HPLC 指紋圖譜

圖2 對照指紋圖譜
2.6 指紋圖譜評價
2.6.1 相似度分析 通過中藥指紋圖譜相似度評價系統(2012 版),對10 批樣品進行相似度評價,S1~S10 相似度分別為 0.980、0.987、0.981、0.990、0.991、0.970、0.979、0.977、0.876、0.983,可知各批樣品質量比較穩定。
2.6.2 主成分分析 通過SPSS 22.0 軟件,以26 個共有峰面積標準化后的數值為變量,計算主成分特征值、累積貢獻率,結果見表1。由此可知,前3 個主成分的累積貢獻率為65.651%,而前5 個為86.042%,由于變量多,故只取前3 個,可代表指紋圖譜共有峰的大部分信息。

表1 主成分特征值及貢獻率
將3 個主成分進行排序,結果見圖3。由此可知,19、14、26、16、25、7、11、3 號峰對主成分1 有顯著貢獻(依次降低),24、23、13、9 號峰對主成分2 有顯著貢獻,6、10 號峰對主成分3 有顯著貢獻,即不同批次樣品的質量差異主要體現在這14 種成分含有量上。其中,14 號峰(芍藥苷)、7 號峰(原兒茶醛)、6 號峰(馬錢苷酸) 為重要參數,并且分別為佐藥白芍、君藥狗脊、君藥續斷的特征性成分。

圖3 3 個主成分排序坐標圖
對主成分載荷值進行計算,得到線性模型,代入各相關數據后得到主成分得分及綜合得分,結果見圖4。由此可知,主成分1 中得分較高的是S2、S4、S6,主成分2 中得分較高的是S5、S7,主成分3 中得分較高的是S3、S4,綜合得分排名依次為S4>S2>S5>S3>S6>S1>S7>S8>S9>S10,其中生產日期較近的批次主成分得分較高,而生產日期較遠的相對較低,即前者質量更好。另外,不同批號樣品中的藥材產地、來源有一定差異,也導致其質量有所不同。
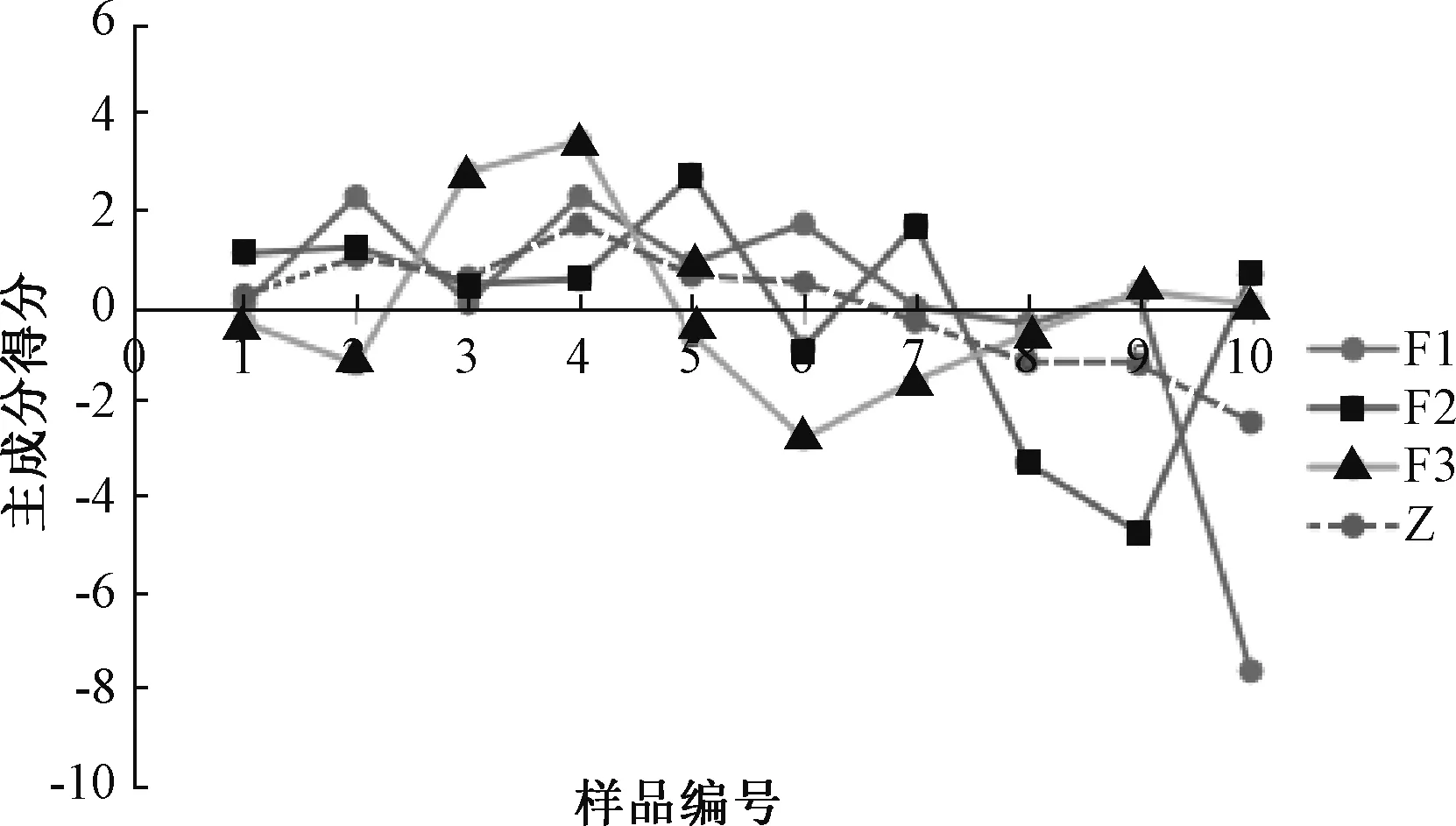
圖4 主成分得分、綜合得分排序坐標圖
3 討論
本實驗篩選提取溶劑時考察了水、甲醇(25%、50%、75%、100%)、乙醇 (25%、50%、75%、100%),發現25%甲醇提取效果最佳,各成分提取均較充分;考察加熱、超聲提取,發現兩者提取效果相當,故選擇更簡單易行的后者;考察了提取時間、料液比,發現兩者分別為30 min、1 ∶20 時提取效率最高;考察了不同流動相比例、色譜柱、體積流量、柱溫,確定“2.1” 項下色譜條件。
然后,建立了10 批尪痹片的HPLC 指紋圖譜,發現有9 批相似度大于0.97,表明其質量比較一致;樣品S9 相似度為0.876,而它為2015 年生產 (有效期至2018 年11月),可能主要是存儲時間較長導致質量有所差異。主成分分析采用降維模式,將反映HPLC 指紋圖譜的多維特征參數用3 個主成分表示,可為尪痹片質量評價提供參考。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
