反應時間對重油懸浮床加氫裂化反應的影響
2020-09-27 09:20:12韓亞鵬楊騰飛鄧文安
石油學報(石油加工) 2020年4期
崔 敏,韓亞鵬,楊騰飛,鄧文安,李 傳
(1.中國石油大學(華東) 理學院,山東 青島 266580;2.中國石油大學(華東) 重質油國家重點實驗室,山東 青島 266580;3.武漢京東方光電科技有限公司,湖北 武漢 430040)
重油懸浮床加氫裂化是一種能夠有效處理高瀝青質、高金屬、高殘炭等重質原料的重油輕質化技術[1-3]。其反應機理為自由基反應機理,類似于在H2存在下的減黏裂化,主要包含裂化反應和縮合反應,在生成具有較高H/C原子比輕質組分的同時,也生成H/C原子比較低的重質產物。這些重質產物容易繼續發生縮合反應而轉變為焦炭[4-7]。重油懸浮床加氫裂化的核心問題就是生焦,尤其是沉積焦的形成。焦炭的生成會導致催化劑失活及管道堵塞,縮短裝置的開工周期[8-9]。
研究人員發現,裂化反應原料中的殘炭含量、氮化物的含量和膠質/瀝青質的組成結構是影響生焦過程的重要因素[10-13]。除原料性質、工藝流程特點和催化劑活性外,反應溫度、反應壓力、反應時間、氫/油比等反應條件也對體系生焦有較大影響。工藝流程的改進及高活性催化劑的開發是解決或減緩生焦問題的常用方法[14-18];合適的反應條件能有效地提高加氫裂化工藝的技術經濟性。
與反應溫度和反應壓力[19]等工藝條件相比,人們對加氫裂化反應時間的研究相對較少,但反應時間不僅能夠決定裝置的處理量,也可用于控制反應深度、調節產物分布及保持催化劑活性[20],是一個極為重要的工藝參數。重油懸浮床加氫技術與其他成熟工藝技術一樣,反應時間短則裝置處理量大、反應深度小,在一定程度上縮短反應時間產生的效果和降低反應溫度的效果基本相同[21]。然而,不同反應時間下重油懸浮床加氫裂化反應的產物分布特點、體系生焦特征、催化劑活性變化情況,以及反應時間的選擇范圍并未被深入了解,其大小的調控更多地是憑借經驗進行。
因此,筆者以委內瑞拉常壓渣油(MRAR)為原料,在高壓攪拌反應釜中研究不同反應時間對重油懸浮床加氫裂化反應產物分布及生焦情況的影響,并采用四組分(SARA)分析法、SEM、XPS等手段對體系膠體穩定性、焦炭形貌、催化劑表面活性金屬的存在形態及相對含量進行了表征分析,以期深入了解反應時間影響重油懸浮床加氫裂化反應特征的因素,為重油懸浮床加氫裂化技術工藝條件的優化提供理論參考。
1 實驗部分
1.1 原料和催化劑
實驗采用MRAR為原料,其主要性質如表1所示。催化劑選取實驗室自制的油溶性環烷酸鉬催化劑,其中金屬Mo質量分數為7.04%。
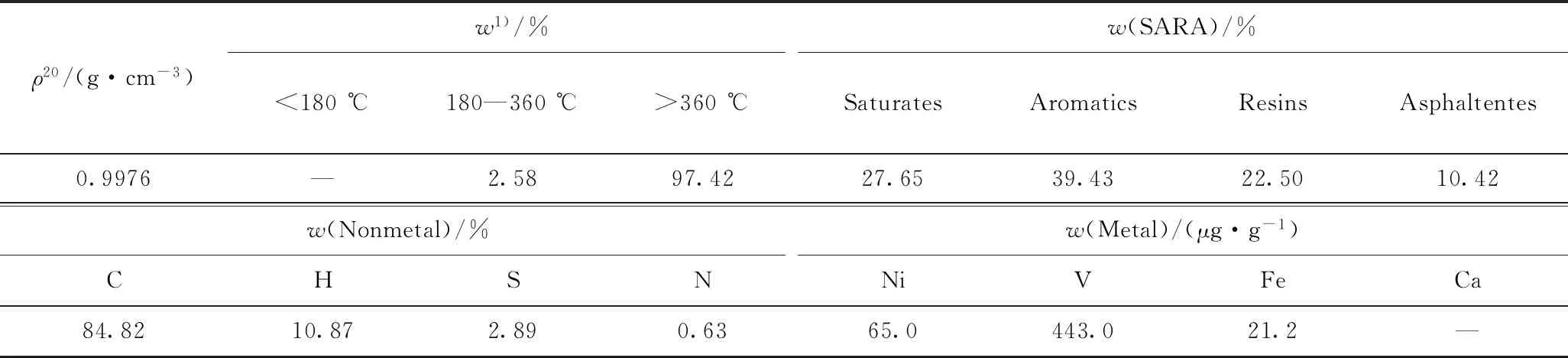
表1 MRAR主要性質和組成
1.2 懸浮床加氫裂化實驗
將150 g的MRAR渣油置于0.5 L高壓釜中,在催化劑質量分數為300 μg/g(按Mo元素計)、反應溫度420 ℃和H2壓力12 MPa條件下,考察反應時間分別為0、30、50、60、70、90、100、110和120 min時MRAR懸浮床加氫裂化反應的產物分布和生焦情況。同時,進行相同條件下的MRAR臨氫熱裂化反應(不添加催化劑)。
1.3 表征
1.3.1 催化劑相對抑制生焦能力
將懸浮床加氫裂化總焦產率與臨氫熱裂化總焦產率進行對比,按式(1)[22]計算催化劑的相對抑制生焦率Cr。
(1)
式(1)中,Cr為催化劑的相對抑制生焦率,%;ys為MRAR懸浮床加氫裂化反應的總焦產率,%;yt為MRAR臨氫熱裂化反應的總焦產率,%。
通過比較不同反應時間Cr值的大小,粗略評價不同反應時間下催化劑的加氫抑制生焦能力的變化。Cr值越大,說明催化劑的相對抑制生焦能力越強。
1.3.2 膠體不穩定性參數(CII)
按照SH/T 0509—92標準將常壓渣油(AR)樣品分離為飽和分、芳香分、膠質和C7-瀝青質四組分(SARA),并根據其含量按式(2)[23]計算AR的膠體不穩定性參數CII。
(2)
式(2)中,wasp、wsat、wres和waro分別為C7-瀝青質、飽和分、膠質和芳香分的質量分數,%。通過CII值近似地反映體系的膠體穩定性情況。CII值越小,表明體系膠體穩定性越好。
1.3.3 焦炭的微觀形貌
將反應生成的懸浮焦和沉積焦研磨成粉末,置于場發射掃描電子顯微鏡(JSM-7500F型)樣品室中,觀察樣品的表面形貌。
1.3.4 催化劑表面Mo元素形態及相對含量
在反應過程中生成的焦炭會逐漸覆蓋催化劑表面的Mo,進而形成懸浮焦和沉積焦。為考察催化劑表面的Mo被焦炭覆蓋的程度,采用Kratos Axis Ultra DLD型多功能電子能譜儀對反應生成的懸浮焦和沉積焦進行XPS能譜分析,并通過XPSPEAK軟件對Mo 3d峰進行分峰擬合[4,24]。
2 結果與討論
2.1 反應時間對MRAR懸浮床加氫裂化反應的影響
2.1.1 反應時間對MRAR懸浮床加氫裂化產物分布的影響
不同反應時間下,MRAR懸浮床加氫裂化產物分布如表2所示。由表2可知:當反應時間為 0 min 時,產物中存在少量氣體、輕油和焦炭,表明反應體系在升溫過程中發生一定的裂化反應;隨反應時間的增加,氣體、輕油和總焦產率均逐漸增加,而未反應的MRAR的比例逐漸減少,說明隨反應時間的延長反應體系中的裂化和縮合反應[21]增多,原料的轉化率和輕質產品、焦炭的產率升高。
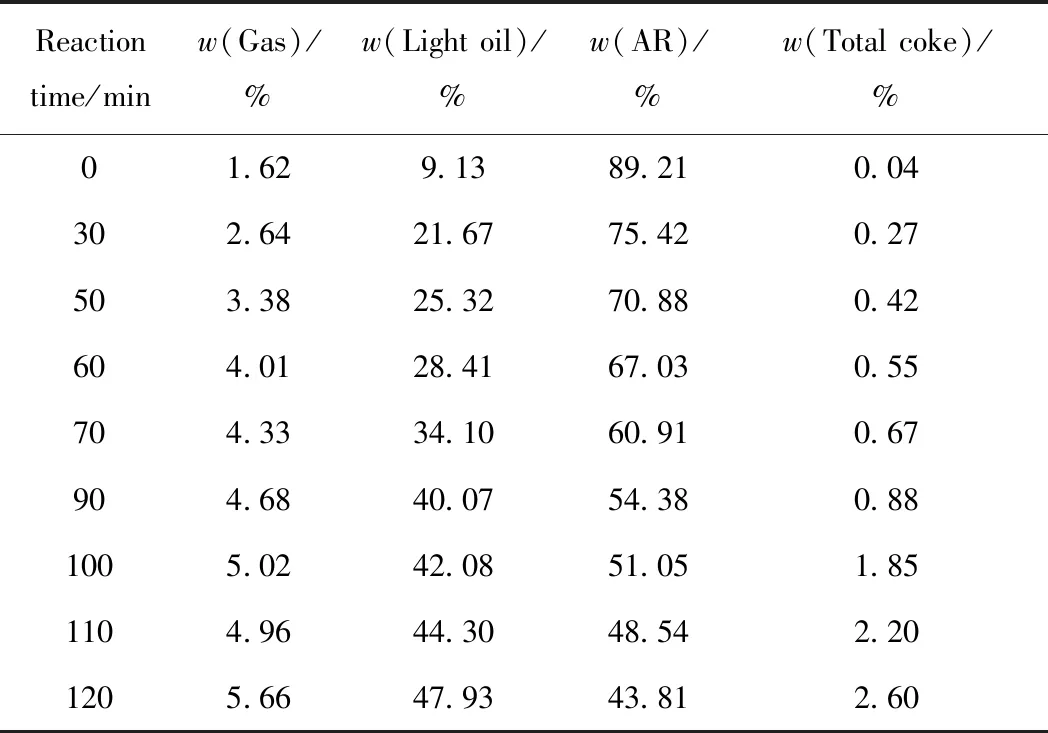
表2 反應時間對MRAR懸浮床加氫裂化反應產物分布的影響
2.1.2 反應時間對反應體系生焦及催化劑相對抑焦率的影響
不同反應時間下,MRAR懸浮床加氫裂化和臨氫熱裂化體系中懸浮焦、沉積焦及總焦產率隨反應時間的變化趨勢見圖1。由圖1(a)可知:MRAR懸浮床加氫裂化反應中,反應時間在70 min內,懸浮焦產率逐漸增加,但總量很低,能夠穩定存在于油相中;在70~90 min時,懸浮焦產率維持基本不變,懸浮焦顆粒變大、發生沉積而轉化為沉積焦[25],沉積焦產率不斷增加;在反應超過90 min后,沉積焦和懸浮焦產率均明顯增加。由圖1(b)可知:MRAR臨氫熱裂化反應中沉積焦與懸浮焦同時生成;隨著反應時間的延長,體系中沉積焦和總焦產率一直增長,而懸浮焦產率先增加后減小,原因在于反應一定時間后,生成的懸浮焦沉積轉變成了沉積焦。此外,臨氫熱裂化的焦炭產率明顯高于懸浮床加氫裂化。
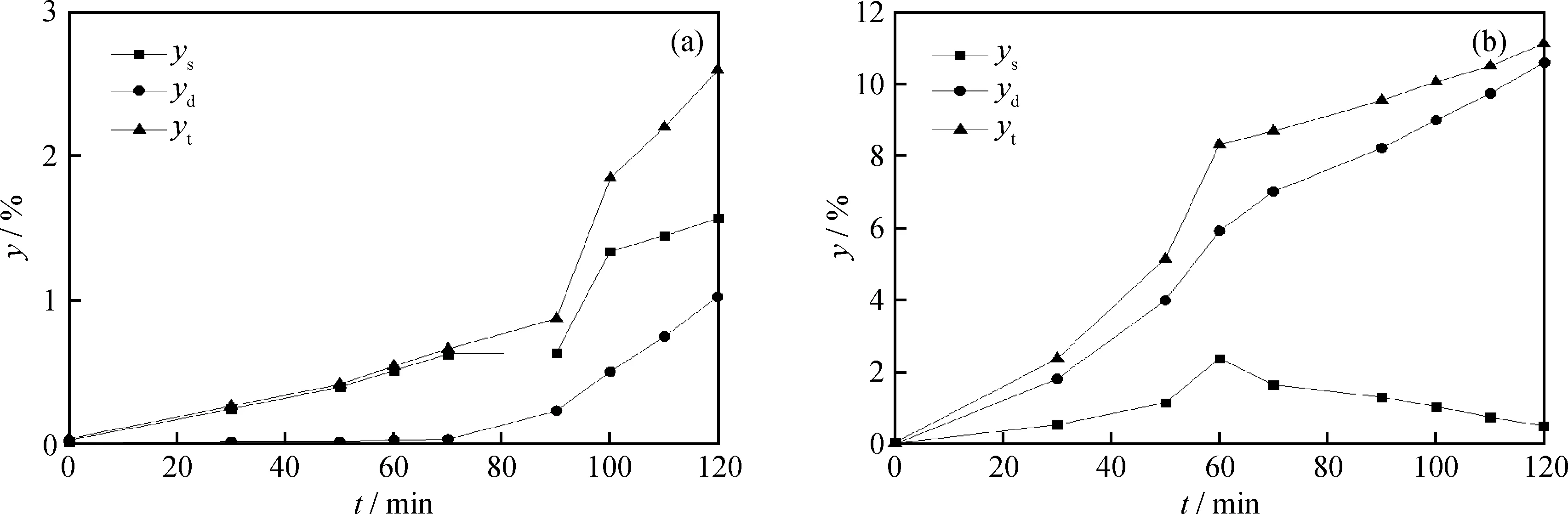
圖1 反應時間對MRAR裂化過程焦炭產率的影響
為了更好地考察催化劑活性隨反應時間的變化情況,按式(1)計算不同反應時間下的催化劑相對抑焦率Cr,結果如圖2所示。由圖2可知,反應初期催化劑抑焦效率較低,主要因為反應初期縮合反應程度較低,焦炭產率較小,并且催化劑中的Mo在升溫過程中的硫化程度不夠完全,使催化劑未能充分發揮其加氫活性。隨著反應時間的延長,在 0~30 min 時,催化劑相對抑焦率迅速上升;在30~60 min時,催化劑相對抑焦率逐漸上升;在反應時間為60 min時達到最高,相對抑焦率達93.41%;反應在60~90 min時,催化劑的相對抑焦率開始緩慢下降;反應90 min后,催化劑相對抑焦率下降較快。這是由于催化劑作為固體懸浮物會成為液相中大分子縮合物的聚集中心,逐漸被大分子包裹形成懸浮焦而覆蓋金屬活性中心,導致催化劑的相對抑焦率下降。
在懸浮床加氫工藝中,沉積焦的生成是造成管道堵塞,開工周期縮短的主要原因之一[26]。根據催化劑在反應過程中的活性變化,優選合適的反應時間,避免大量生成沉積焦,對懸浮床加氫工藝的長周期運轉具有重要意義。因此,按照體系催化劑相對抑焦能力、體系總焦和沉積焦產率的變化,MRAR懸浮床催化加氫裂化反應優化的反應時間為90 min。此時,體系的總焦和沉積焦產率分別為0.88%和0.24%。
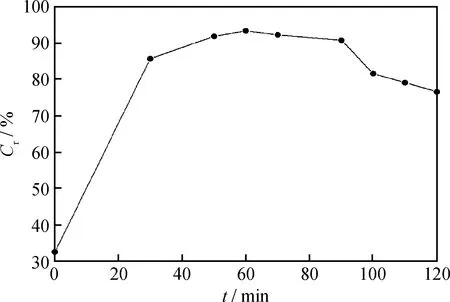
圖2 反應時間對催化劑相對抑焦率的影響
2.2 反應時間影響MRAR懸浮床加氫裂化反應的機理
2.2.1 不同反應時間下體系的膠體穩定性
不同反應時間下MRAR加氫裂化尾油的SARA分析結果見表3。由表3可知,隨反應時間的延長,尾油中飽和分和瀝青質含量逐漸增加,芳香分含量減少,膠質含量沒有明顯規律性的變化,但芳香分和膠質的含量之和持續降低。作為分散相的瀝青質相對含量增加,導致體系大分子團聚的傾向增加;而作為溶劑的膠質和芳香分的含量減少,使瀝青質膠束外圍的溶劑化層減弱,體系的膠體穩定性變差。

表3 不同反應時間下加氫裂化尾油四組分組成
根據表3中SARA數據,由式(2)計算反應體系膠體的不穩定性參數CII值,結果如圖3所示。由圖3可知:反應時間在60 min內CII值較低,且基本保持穩定,表明此階段體系膠體穩定性較好,生成的懸浮焦能穩定地懸浮于油相中,沒有明顯轉化為沉積焦的現象;在60~90 min時,CII值逐漸升高,表明體系的膠體穩定性逐漸降低,溶焦能力達到臨界點,懸浮焦逐漸轉化為沉積焦;當反應時間超過90 min后,CII值升高趨勢更明顯,表明反應體系膠體穩定性急劇下降,沉積焦和懸浮焦產率快速上升。隨反應時間的延長,CII值的變化趨勢與焦炭產率的變化趨勢基本一致,說明反應時間對反應體系膠體穩定性的影響是其對MRAR懸浮床加氫裂化反應影響的特征之一。
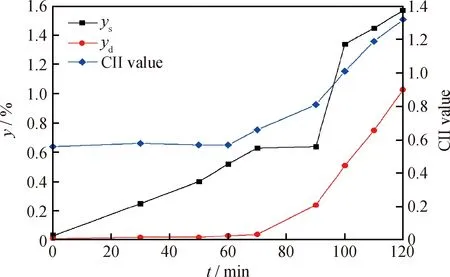
圖3 不同反應時間下MRAR懸浮床加氫裂化體系的CII值及焦炭產率
2.2.2 不同反應時間下焦炭的形貌特征
不同反應時間下懸浮焦的微觀形貌分別如圖4所示。由圖4可知:反應時間在30 min時,體系受熱時間較短,不易生焦,生成的懸浮焦為細小的片狀結構,邊緣較薄,中間有較大的孔隙,結構比較疏松;50 min時,懸浮焦片狀結構變大,結構緊密,片狀邊緣開始翹起;60 min時,懸浮焦開始呈現為細小球狀顆粒焦,其產率逐漸增加;70 min時,炭質微球數量較多,直徑約為1 μm,表面粗糙;90 min時,由于體系的膠體穩定性變差,溶焦能力達到臨界點,懸浮焦炭質微球開始轉化為沉積焦;110 min時,懸浮焦小球變大,并出現合并現象。

圖4 不同反應時間下的懸浮焦SEM照片
沉積焦的微觀形貌如圖5所示。由圖5可知,與懸浮焦相比,沉積焦的結構更致密。在反應時間為70 min時,出現直徑約0.4 μm的沉積焦小球,且片狀結構呈花瓣狀向內彎曲,球狀顆粒有繼續生長的趨勢;反應在90~110 min時,沉積焦含量逐漸增加,此時生成的小球逐漸變得光滑;反應在110 min時,沉積焦小球的直徑約為1.5 μm。這說明在沉積焦含量增加的同時碳質小球的粒徑也在不斷增加,懸浮焦向沉積焦轉化的同時,沉積焦中的小球自身也在不斷生長。

圖5 不同反應時間下的沉積焦SEM照片
2.2.3 不同反應時間下催化劑表面Mo的形態及相對含量
為了解催化劑表面活性金屬Mo在不同反應時間下的存在形態及相對含量,對不同反應時間的懸浮焦和沉積焦進行了XPS分析。以反應時間為 60 min 時懸浮焦和70 min時沉積焦為例,其XPS全掃描譜圖及Mo 3d峰的分峰擬合圖見圖6。由圖6(a)和(c)可知,懸浮焦和沉積焦表面均含有C、O、S、N和Mo元素,說明活性金屬Mo并未被焦炭完全覆蓋,催化劑表面仍具有加氫抑焦活性。由Mo 3d 峰擬合結果(圖6(b)和(d))可知,催化劑表面的Mo存在Mo4+和Mo6+等多種形態,其擬合峰強度存在明顯差別。不同反應時間下帶懸浮焦和沉積焦的催化劑表面Mo的相對含量見表4。由表4可知:隨反應時間的延長,催化劑表面的Mo元素相對含量均逐漸下降,說明催化劑表面焦炭的覆蓋率逐漸增加;反應時間在70 min內,催化劑表面的Mo元素相對含量下降的幅度較小;而超過70 min后,其下降趨勢明顯增強,同時體系出現明顯的沉積焦,說明反應70 min后,催化劑表面懸浮焦覆蓋率明顯增加,進而發生沉積形成沉積焦。
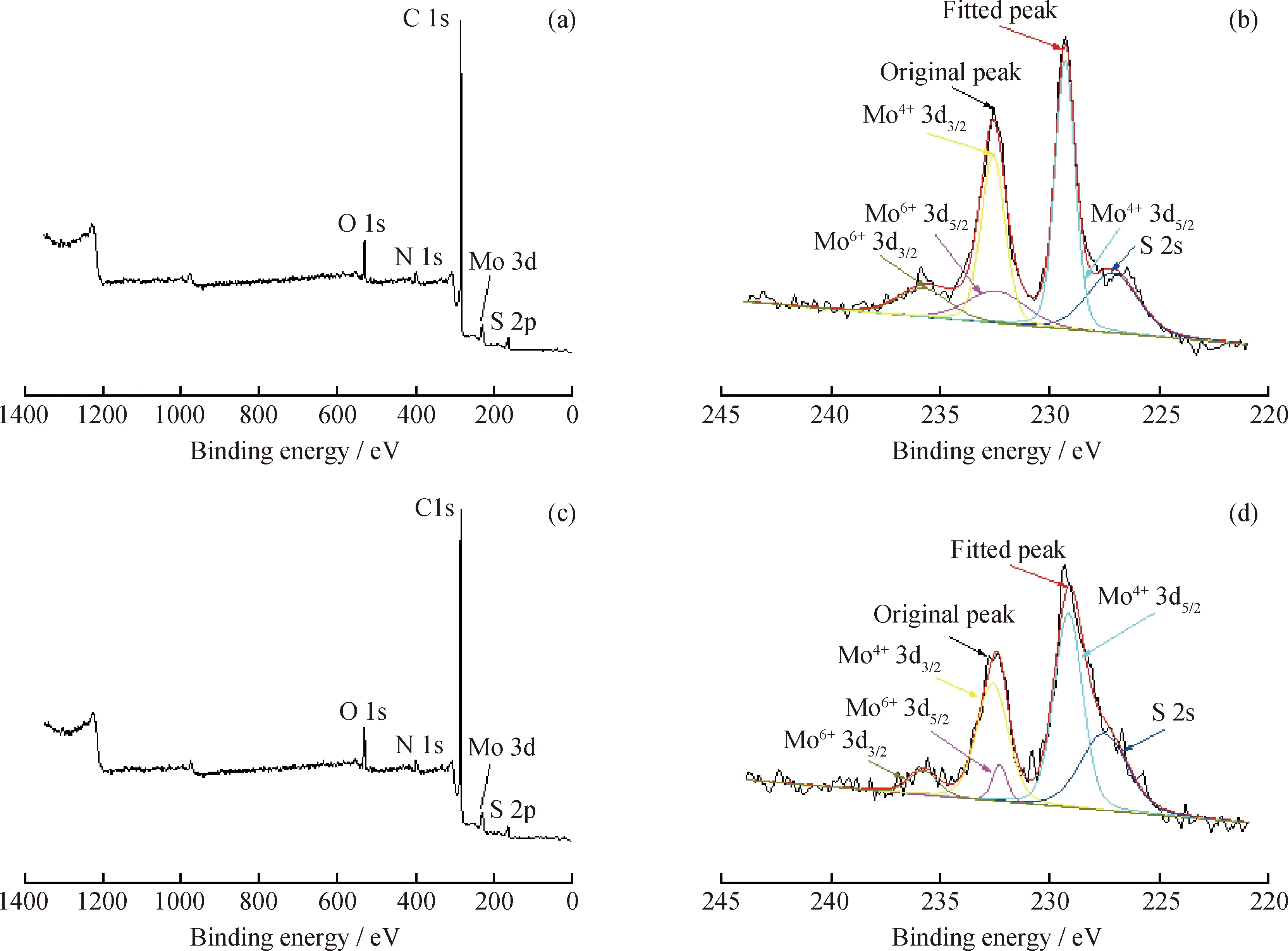
圖6 反應在60 min時懸浮焦和70 min時沉積焦的XPS譜圖
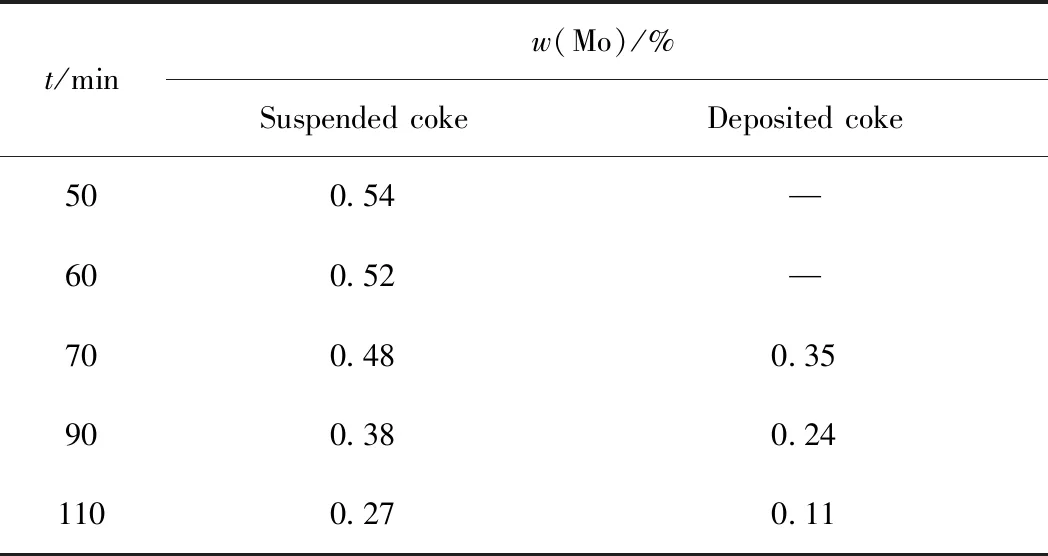
表4 不同反應時間下覆蓋懸浮焦和沉積焦的催化劑表面的Mo質量分數
通過對Mo 3d峰的分峰擬合峰面積的計算,可得催化劑表面不同形態Mo的相對含量,結果列于表5。由表5可知:隨反應時間的延長,催化劑表面的Mo4+相對含量均逐漸增加,而Mo6+相對含量逐漸減小;反應時間在70 min內,Mo4+和Mo6+相對含量變化的幅度均很小;超過70 min后,二者的變化趨勢明顯增強。此外,與相同反應時間下的懸浮焦相比,沉積焦Mo4+的相對含量略高,Mo6+的相對含量則略低。
Mo4+相對含量更高,表明催化劑表面Mo的主要存在形態為MoS2,其具有很高的加氫抑焦活性[27-28];而Mo6+可歸于MoS3的生成及測試過程中樣品表面的氧化等。焦炭在形成過程中較難生長在高活性的Mo4+位,會更傾向于在Mo6+位聚集形成懸浮焦[29],因而隨反應時間延長,催化劑表面懸浮焦覆蓋的Mo6+位逐漸增多,因而測得Mo4+相對含量上升;Mo6+相對含量下降。當催化劑表面上覆蓋足夠多焦炭時,懸浮焦沉積轉變為沉積焦。此時,催化劑表面的Mo6+位覆蓋的焦炭相對較多,表現為在覆蓋沉積焦催化劑上的Mo6+相對含量比在覆蓋懸浮焦催化劑上的略少,且有隨反應時間延長繼續下降的趨勢。
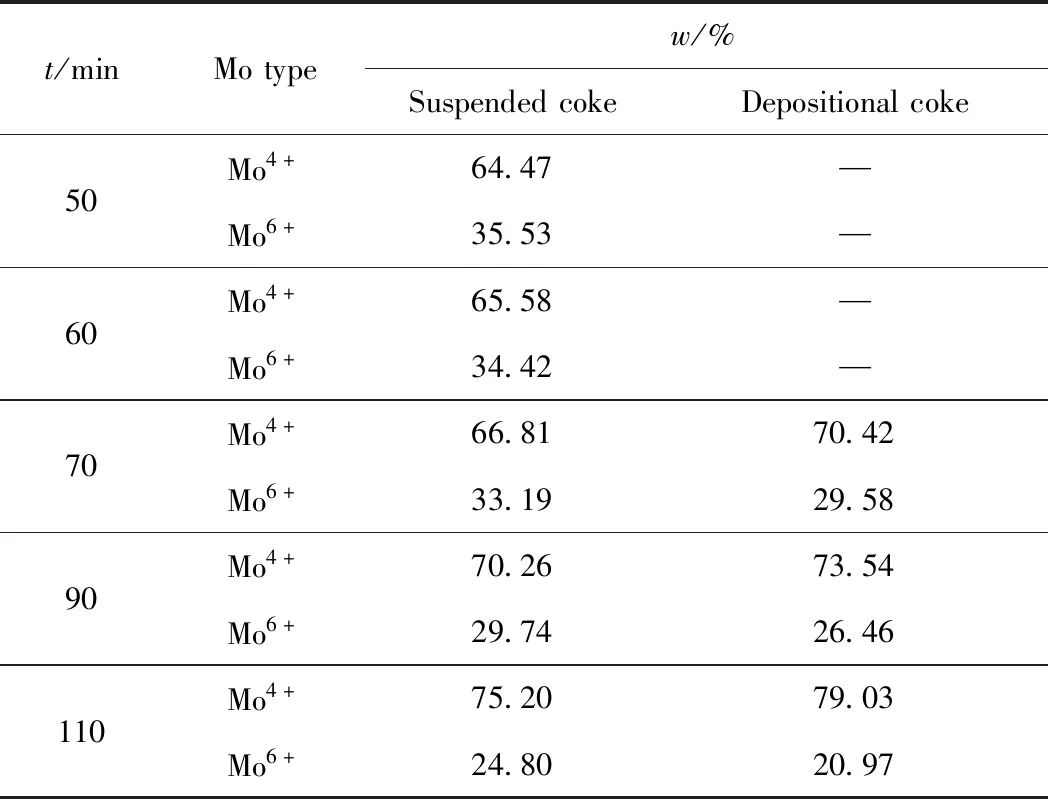
表5 不同反應時間下催化劑表面Mo形態及相對含量
3 結 論
(1)當MRAR懸浮床加氫裂化反應時間延長時,原料的轉化率和輕質產品、總焦炭的產率升高。反應在70 min時,懸浮焦開始轉化為沉積焦,然后二者的量不斷增加。隨著裂化反應的進行,催化劑相對抑焦率不斷提高;在反應60 min時達到最高,然后緩慢下降;在90 min后相對抑焦率變差。因此,MRAR懸浮床加氫裂化反應時間以90 min內為宜。
(2)反應過程中,反應體系中膠體穩定性、催化劑表面Mo元素含量及焦炭產率變化趨勢一致,說明反應時間對MRAR懸浮床加氫裂化反應的影響主要在體系膠體穩定性變化和焦炭覆蓋催化劑活性金屬的程度等方面。
(3)隨反應時間延長,表面覆蓋焦炭的催化劑上Mo6+相對含量降低,Mo4+相對含量升高。其原因在于裂化反應生成的焦炭更易覆蓋在催化劑表面的Mo6+位上。
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應用化工(2014年3期)2014-08-16 13:23:50
