均苯三甲酸過渡金屬配位聚合物的合成、表征及性質研究
2020-09-29 06:54:58王潤雪喬勁松李紅雙
科學技術創新 2020年29期
關鍵詞:實驗
王潤雪 高 巍 喬勁松 李紅雙
(吉林建筑科技學院,吉林 長春130114)
金屬有機骨架材料是一種呈三維網絡結構的新型多孔材料,主要以均苯三甲酸作為配體,由均苯三甲酸根離子與金屬離子連接組成其骨架結構,在催化、氫分離、儲氫等領域具有較大的開發潛力,可為工業生產及日常生活提供更加理想的多孔材料。
1 配位聚合物
1.1 組成結構
配位聚合物通常以金屬、金屬簇作為連接器,以有機配體作為連接體,依據配位鍵分為一維、二維、三維等結構類型,其中典型的一維包含直鏈、雙鏈、螺旋鏈等,二維結構體現為四方格子形網絡,三維結構則包含金剛石拓撲網絡、石英、方硼石等多種復雜結構[1]。
1.2 多孔配位聚合物
相較于常規配位聚合物,多孔配位聚合物在氣體催化、儲存與分離上具備更好的性能優勢。常用配體材料分為含N 給體的配位、含O 給體的配位兩種類型,前者多選用剛性中性配體,孔道有限;后者在配位后配體呈負電性,骨架多為中性。基于此,擬采用羧配類配體與過渡金屬合成配位聚合物,用于判斷不同合成條件對于最終配位聚合物骨架穩定性的影響[2]。
2 基于溶劑熱法合成均苯三甲酸及其表征、性質分析
2.1 均苯三甲酸- 微孔鋅
2.1.1 實驗操作
實驗儀器包含STA409PC 綜合熱分析儀、SMART APEX Ⅱ型單晶X- 射線衍射儀,實驗工具為高壓釜和烘箱,實驗試劑選用均苯三甲酸、Zn(CH3COO)2·2H2O、N,N- 二甲基甲酰胺等。量取0.26g 濃 度 為1mmol 的Zn (CH3COO)2·2H2O 和0.25g 濃 度 為1.2mmol 的均苯三甲酸置于高壓釜內,向高壓釜內加入10mL 的N,N- 二甲基甲酰胺,在140°C 溫度條件下加熱48h 后取出,待冷卻至室溫后生成透明塊狀晶體,利用N,N- 二甲基甲酰胺清洗3 次后進行過濾、烘干保存,產率約為84%。取規格為0.36mm×0.3mm×0.2mm 的透明塊狀晶體放入衍射儀內,利用經由石墨單色器處理后的Mo 靶Kα 射線在區間 (2.26°,28.29°)內進行掃描,在293(2)K 下收集到3813 個衍射點,最終偏差因子R1=0.062、wR2=0.118。基于計算機Shelxtl 軟件程序進行數據的經驗吸收校正,利用直接法完成晶體結構的測定,借助最小二乘法完成坐標與參數的修正。
2.1.2 晶體結構分析
通過觀察生成的配位聚合物Zn(C3Cl6O3)·NH2(CH3)2·HCON(CH3)2的晶體結構特征可知(其分子片斷如圖1 所示),該晶體呈三維結構、P21/n 空間群,由二聚鋅簇與均苯三甲酸構成配位聚合物的分子結構。在二聚鋅簇中Zn 原子均與個四羰基O 原子配位,組成畸變的四面體;在均苯三甲酸配體中,1 個羰基與2個Zn 原子間基于螯合作用完成配位,其余2 個羰基則與1 個Zn 原子配位,由此組成三維網絡結構。將該晶體結構進行簡化,可將二鋅簇、均苯三甲酸分別視為畸變八面體與三棱錐,進而將整體三維網絡結構近似為二氧化鈦拓撲網絡結構,其中的鋅二簇分別與6 個均苯三甲酸配位,均苯三甲酸分別與3 個鋅二簇配位。
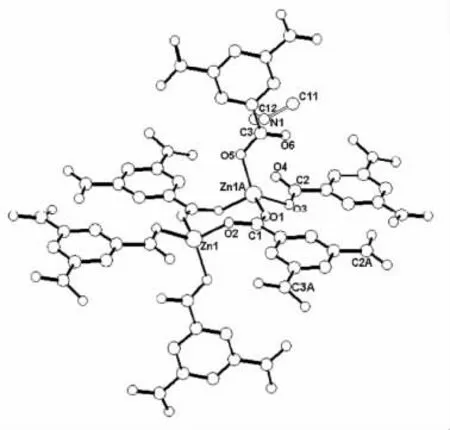
圖1 配位聚合物分子片斷圖
2.1.3 聚合物性質分析
在熱穩定性分析上,該配位聚合物主要經歷以下三個反應階段:其一是由110°C 升至170°C,在此期間配位聚合物開始分解,失重率為18.89%,說明各分子式中均失去一個N,N- 二甲基甲酰胺分子;其二是由170°C 升至270°C,在此期間配位聚合物無明顯失重現象;其三是待反應溫度升高至270°C 以上后,配位聚合物的骨架開始分解,說明客體N,N- 二甲基甲酰胺分子較容易失去,但其隧道體積占比仍保持在三分之一左右。
為進一步檢驗該骨架的穩定性與材料的多孔性,取配位聚合物樣品置于烘箱內加熱至100°C 并保持6h,隨后將生成的樣品進行稱重,失重率為18.72%,說明N,N- 二甲基甲酰胺分子已全部失去,此時觀察衍射譜圖顯示產物仍保持晶態,但因化合物骨架變形導致其衍射峰朝向高角度移動;接下來再將樣品置于N,N- 二甲基甲酰胺溶劑中浸泡24h,此時觀察衍射譜圖可發現其偏移的衍射峰恢復至原位,說明配位聚合物在重新吸附N,N- 二甲基甲酰胺分子后其骨架變形得以恢復,與推斷結果相符。
2.2 均苯三甲酸- 鎘配位聚合物
2.2.1 實驗操作
實驗儀器包含Avatar360 型FT-IR 紅外光譜儀、STA409PC綜合熱分析儀、SMART APEX Ⅱ型單晶X- 射線衍射儀與CHI660D 電化學工作站,實驗工具包含25mL 反應釜、烘箱等,實驗試劑選用均苯三甲酸、硝酸鎘、N,N- 二甲基甲酰胺作為分析純。量取1mmol 均苯三甲酸、1.5mmol 置于反應釜中,依次加入7mL 的N,N- 二甲基甲酰胺和水,將混合物送入烘箱內在120°C 溫度下反應24h 左右,隨后取出進行冷卻處理,可觀察到內壁分布有透明狀晶體,取1~2 塊晶體進行X- 射線衍射處理,收集剩余晶體經過濾、烘干后保存,產率約為63%。取規格為0.3mm×0.25mm×0.2mm 的透明塊狀晶體放入衍射儀中,利用經石墨單色器處理后的Mo 靶Kα射線在區間 (2.76°,29.06°)內進行掃描,在293K 下收集到的衍射點共計3213 個,其中包含2825 個獨立衍射點(Rint=0.0585),最終偏差因子R1=0.078、wR2=0.185。基于計算機Shelxtl 軟件程序進行數據的經驗吸收校正,利用直接法完成晶體結構的測定,借助最小二乘法完成坐標與參數的修正[3]。
2.2.2 晶體結構分析
通過觀察生成的鎘配位聚合物{[Cd3(C9H3O6)2(H2O)9]·2H2O}n的晶體結構特征可知(其分子片斷如圖2 所示),該晶體呈單斜結構、C2/c 空間群,均苯三甲酸的3 個羧基與稀有金屬鎘的3個Cd(Ⅱ)在螯合作用下完成配位,構成配位聚合物的分子結構。選取生成的鎘配位聚合物的分子片斷進行觀察,其中Cd1(Ⅱ)與鄰近的7 個O 原子配位組成雙五角錐形的分子結構,其中1a、2a、3、4、8 五個O 原子基本保持共面,7、9 兩個O 原子則分別位于S、N 兩極;Cd2(Ⅱ)與鄰近的7 個O 原子配位組成畸變的單帽三棱柱結構,其中5b、6b、10、11b、11、6 六個O 原子分別位于三棱柱的兩面,O 原子5 則位于平面外側的帽位置。從中可以看出,Cd1(Ⅱ)周圍分布的離子主要通過均苯三甲酸實現連接,均苯三甲酸即為橋聯配體,以a 軸為基準無限延伸構成一維鏈;Cd2(Ⅱ)周圍分布的離子與2 個均苯三甲酸的不同羧基連接配位,并且與Cd1(Ⅱ)形成的一維鏈連接,由此使配位聚合物的分子結構在ac 面、ab 面分別呈現為夾心結構和棱狀結構。
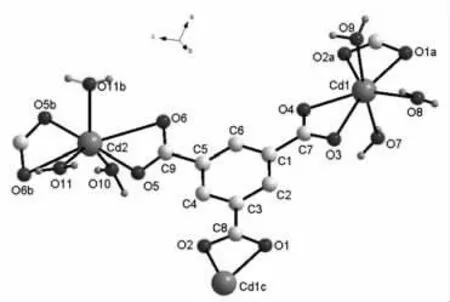
圖2 配位聚合物分子片斷圖
2.2.3 聚合物性質分析
在紅外光譜分析上,選取KBr 壓片法獲取配位聚合物在(4000cm-1,400cm-1)區間內的紅外光譜,可觀察到羧基在1711cm-1處的不對稱伸縮振動吸收峰呈消失狀態,羧基在1615cm-1、1372cm-1兩處分別出現不對稱伸縮振動吸收峰與對稱伸縮振動吸收峰。針對羧基產生位移的原因進行分析,主要體現為其中的O 原子參與到配位過程中,O 原子的弧對電子朝向金屬離子靠攏,羰基的鍵力常數減小,進而導致羧基的可觀測頻率縮小[4]。
在熱穩定性分析上,選取Al2O3作為參考物質,將Al2O3與配位聚合物分別置于綜合熱分析儀中以10°C/min 的升溫頻率進行測定。通過觀察熱重曲線可知,配位聚合物的反應過程主要分為兩個階段:其一是反應溫度由90°C 升高至150°C 時,配位聚合物逐步開始分解,失重率為22.14%;其二是反應溫度由150°C 升高至712°C 時,配位聚合物中均苯三甲酸的骨架逐步加快分解,待溫度上升至712°C 后其失重率基本不再發生變化,剩余產物重量約為22.18%,可推斷出產物為CdO。
在電化學性質分析上,選取玻璃碳、鉑、甘汞電極構成三電極體系,將N,N- 二甲基甲酰胺與H2O 作為溶劑,配制出濃度為1.0×10-5mol/L 的配位聚合物,選取濃度為2.0mol/L 的NaCl 溶液作為電解質。以N2氣體作為測定環境,在(-1.5V,0.8V)區間內掃描配位聚合物,生成其電化學曲線。以N,N- 二甲基甲酰胺作為空白對照組,觀察配位聚合物的電化學曲線可知,配位聚合物在-0.8V 處出現一尖峰,與電對Cd(Ⅱ)發生電子轉移相對應,其電流值為5.694×10-5A,由此說明在該電解過程中發生的電子轉移為不可逆的。
3 結論
總體來看,本文基于溶劑熱合成法開展了2 個關于均苯三甲酸過渡金屬配位聚合物的實驗,在均苯三甲酸- 微孔鋅實驗中獲得了一個由畸變的八面體與三棱錐組成的三維網絡結構,近似二氧化鈦拓撲結構,整體結構具有較強的穩定性、骨架具有多孔性;在均苯三甲酸- 鎘配位聚合物實驗中獲得了一個由變形的五角雙錐體與畸變的單帽三棱柱體組成的三維網絡結構,在電解過程中電子轉移為不可逆的。由此說明,將均苯三甲酸作為配體,能夠有效構筑出具有多孔骨架的金屬配位聚合物,對于金屬有機骨架材料的合成具有重要研究價值。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
