Fe3O4@SiO2-NH2粒子對鈾(Ⅵ)在阿拉善水相中的吸附性能研究
2020-10-13 00:52:16王彥惠冷陽春成建峰衛純純趙玉婷李東瑞
核科學與工程 2020年4期
王彥惠,冷陽春,成建峰,衛純純,趙玉婷,李東瑞
(1.成都理工大學 核技術與自動化工程學院,四川 成都 610000;2.西南科技大學 國防科技學院,四川 綿陽 621010)
目前,在國際能源短缺和低碳經濟的推動下,核能成為能源發展的主流趨勢。中國已成為核能開發利用的主要國家,但利用核電站發電的同時產生了大量的核廢料[1]。作為主要的核燃料,鈾是工業、農業、軍工和航空航天工業的基礎能源之一,其開采和加工不可避免地產生大量含U(Ⅵ)的放射性廢水[2]。從核工業中泄漏的U(Ⅵ),能夠引起地表水和地下水的污染,這引起了全球的廣泛關注。內蒙古阿拉善地區作為我國高水平放射性廢物地質處置庫預選場址之一,其阿拉善黏土巖性能和地下水環境是研究的重點。
內蒙古阿拉善黏土巖是高放廢物地質處置庫的重要候選圍巖之一,與自然界其他吸附材料相比具有自封閉性、滲透率低等的優點[3]。但是黏土巖作為吸附劑受特定環境的限制,對放射性核素的吸附效果不夠理想,因此,近年來磁性納米粒子因其獨特的磁性和制備的可行性引發了各國學者的極大興趣。徐要輝等[4]采用溶膠—凝膠法制備了氨基功能化的Fe3O4@SiO2復合微球,實現了對Pb2+、Cr3+、Cd2+和Hg2+的良好富集和分離;尹甲興等[5]制備出氨基化磁性粒子Fe3O4@SiO2-NH2,并研究其作為吸附劑在不同條件下對Pb2+的吸附性能,結果表明所制備的復合粒子對Pb2+有較大的吸附容量,可作為有效處理含鉛廢水的吸附材料。
本實驗利用正硅酸乙酯(TEOS) 和3-氨丙基三乙氧基硅烷(3-APTES)的水解在Fe3O4顆粒表面進行包裹和氨基接枝,制備出一種磁性納米吸附劑,以阿拉善地下水為水相環境研究對U(Ⅵ)的吸附性能,同時加入阿拉善黏土巖吸附實驗組與磁性納米材料進行對比。
1 材料與方法
1.1 儀器與試劑
1.1.1 重要儀器
DZF-6000型真空干燥箱;CHA-SA氣浴恒溫振蕩器;JJ-2型控溫電動攪拌器;UPT-Ⅱ-10T型純水機;ISO—9001型高壓反應釜;Aiglent1200/7700x型電感耦合等離子體發射光譜-質譜儀(ICP-MS)。
1.1.2 試劑
FeCl3·6 H2O,無水CH3COONa,氨水,乙二醇,聚乙二醇,正硅酸乙酯(TEOS),3-氨丙基三乙氧基硅烷(3-APTES),U3O8,無水乙醇等均為分析純,阿拉善地下水。阿拉善地下水成分分析[6]如表1所示。
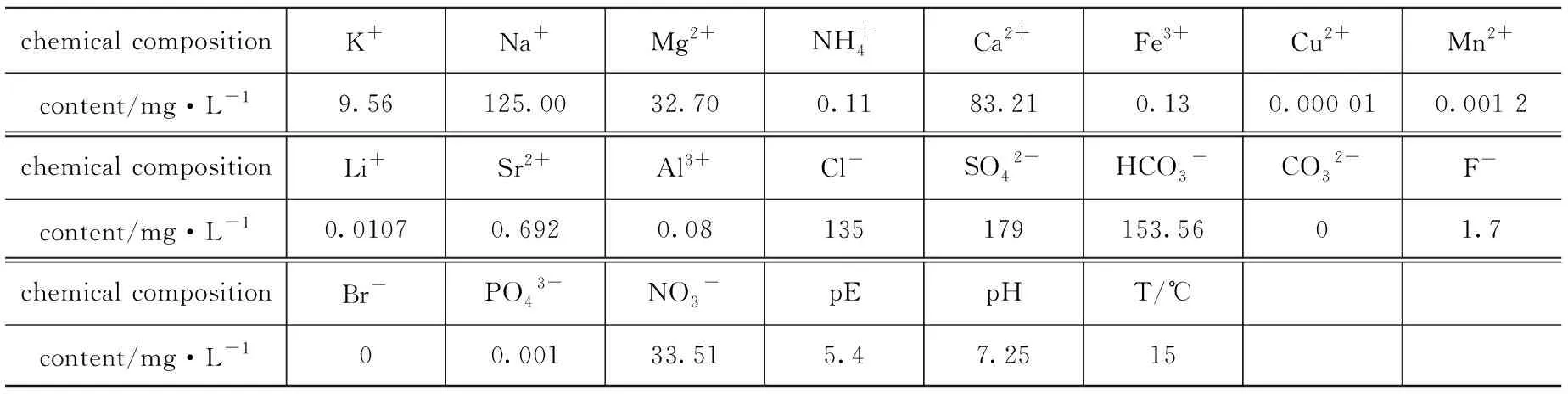
表1 阿拉善某地下水成分分析Table1 Composition analysis of Alashan groundwater
1.2 Fe3O4@SiO2-NH2磁粒子的制備
根據徐要輝等[4]和王彥惠等[7]的方法,利用溶劑熱法制備出分散良好的Fe3O4顆粒,再用TEOS和3-APTES依次對Fe3O4顆粒表面包覆與修飾,制得Fe3O4@SiO2-NH2磁性納米材料。
1.3 吸附實驗
室溫下,分別稱取0.04 g的Fe3O4@SiO2-NH2粒子和阿拉善黏土巖于10 mL聚丙烯離心管中,加入7.2 mL阿拉善地下水,再加入0.8 mL 200 μg·mL-1的U(Ⅵ)標液進行震蕩,達到吸附平衡時間后離心分離固液相,吸取上清液稀釋后利用ICP-MS設備[8]測定。探究接觸時間、pH、固液比和溫度對吸附的影響。
1.4 數據處理
吸附率E%計算公式如下:
(1)
吸附容量qt/μg·g-1計算公式如下:
(2)
平衡吸附容量qe/μg·g-1計算公式如下:
(3)
式中:C0——U(Ⅵ)的初始濃度,μg·mL-1;
Ct——t時測得U(Ⅵ)的濃度,
μg·mL-1;
Ce——吸附平衡時測得U(Ⅵ)的濃度,μg·mL-1;
m——磁性納米粒子的質量,g;
V——吸附實驗中液體總體積,mL。
2 結果與分析
2.1 表征分析
2.1.1 掃描電子顯微鏡(SEM)分析
圖1為Fe3O4@SiO2-NH2磁性納米粒子的掃描電鏡。
從圖1可以看出,Fe3O4@SiO2-NH2是規則的球形體,粒徑約200 nm,納米Fe3O4被包覆和修飾,分散良好。該結構闡明了Fe3O4顆粒被致密的SiO2良好包裹而不是物理粘附或混合到SiO2中,這使得磁性顆粒在酸性環境中更穩定。因此,在Fe3O4表面上用SiO2包裹和與氨基的接枝可以有效地消除納米Fe3O4的團聚,這有利于吸附活性位點的暴露,與溶液中的U(Ⅵ)完全接觸,提高吸附性能。

圖1 Fe3O4@SiO2-NH2磁性納米粒子的SEM圖Fig.1 SEM diagrams of Fe3O4@SiO2-NH2 Composite Magnetic Nanoparticles
2.1.2 透射電鏡(TEM)分析
圖2為磁性納米粒子的TEM圖。
圖2為200nm標尺下Fe3O4@SiO2-NH2磁性納米粒子的透射電鏡圖,由圖可知,粒徑均勻,大約為200 nm,和掃描電鏡結果相符合。同時粒子具有明顯的核殼結構,從圖中可以看到內層為深色的Fe3O4粒子,外面包圍著一層非晶態的物質,即為SiO2,且包覆層致密完整,具有更好的環境穩定性,可以在酸性環境中保持穩定。

圖2 Fe3O4@SiO2-NH2納米磁粒子的TEM圖Fig.2 TEM diagrams of Fe3O4@SiO2-NH2 Composite
2.1.3 X射線衍射儀(XRD)分析
圖3為Fe3O4@SiO2-NH2磁性納米粒子的XRD圖。
圖3分別顯示為裸Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2-NH2的XRD圖譜,從圖譜可明顯觀察到位于2θ=30.08°、35.46°、43.08°、53.48°、56.98°和62.58°的衍射峰,分別對應Fe3O4的(220)、(311)、(400)、(422)、(551)和(440)晶面。裸Fe3O4顆粒和Fe3O4@SiO2-NH2中的衍射峰的峰行和峰寬基本一致,均為Fe3O4粒子的衍射峰,表明在修飾后Fe3O4的晶體結構得到了很好的保持。包裹后的粒子在20°~30°內出現了較大的波包[9,10],這代表無定型SiO2的存在。Fe3O4@SiO2-NH2衍射峰與其他兩者比較發生了較明顯的不同,說明粒子表面氨基改性的成功。
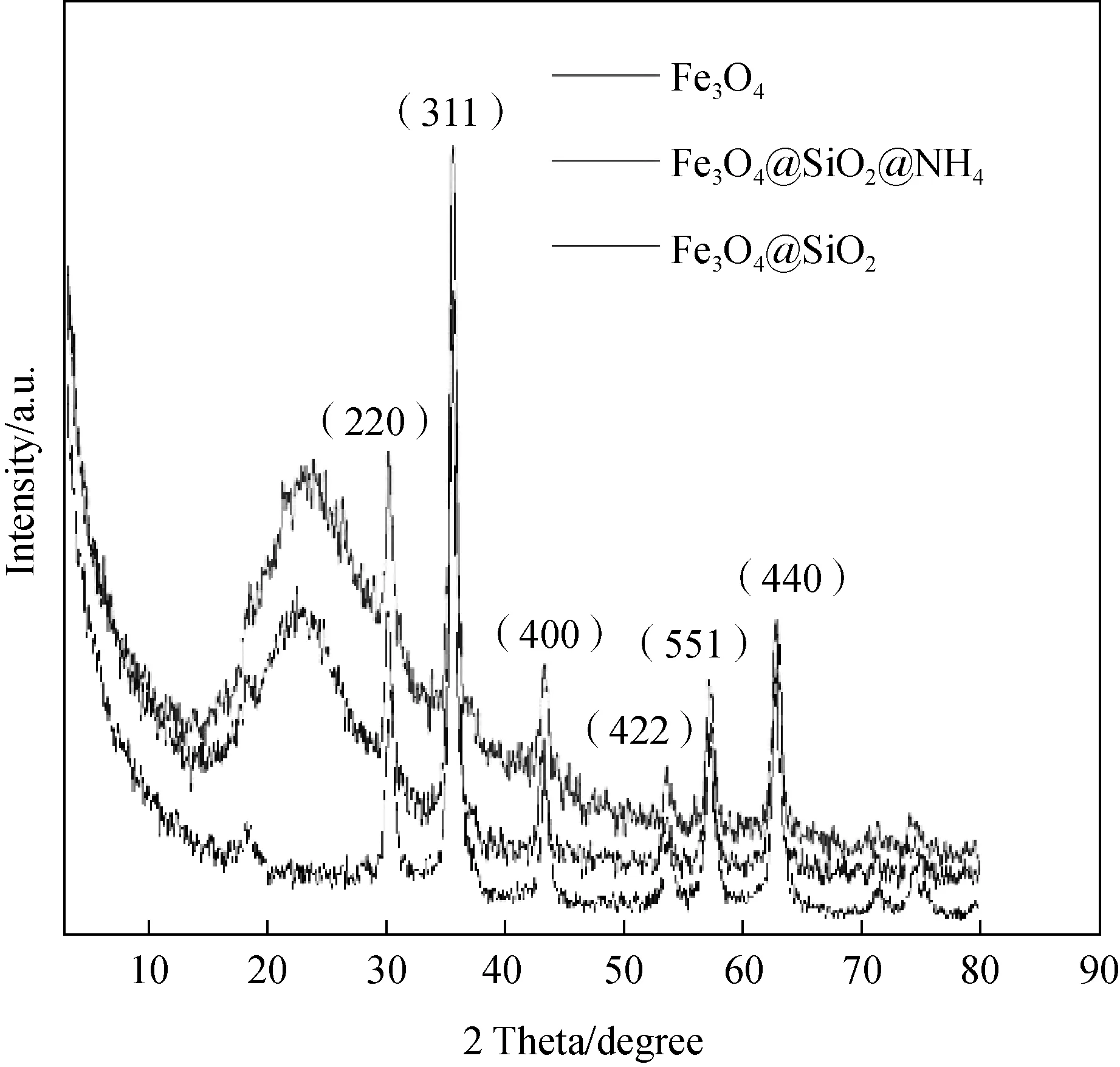
圖3 Fe3O4@SiO2-NH2納米磁離子的XRD圖Fig.3 XRD Charts of Fe3O4@SiO2-NH2 Composite Magnetics Nanoparticles
2.2 吸附劑對鈾的吸附性能研究
2.2.1 接觸時間對U(Ⅵ)在吸附劑上吸附特征的影響
圖4顯示了接觸時間對U(Ⅵ)吸附到吸附劑上的影響。復合粒子在120 min達到平衡,阿拉善黏土巖在20 h達到吸附平衡,且復合粒子的平衡吸附率遠大于阿拉善黏土巖。根據上述結果,將振蕩時間分別固定130 min和24 h以確保完全達到平衡。在這種條件下吸附U(Ⅵ)的動力學由兩個階段組成:初始快速階段和較慢的第二階段。初始階段的快速去除率歸因于U(Ⅵ)快速與附劑的外表面的活性位點組裝[11,12]。較慢階段由于濃度梯度降低并且可用的自由活性位點達到飽和,導致后期的吸附速率降低。
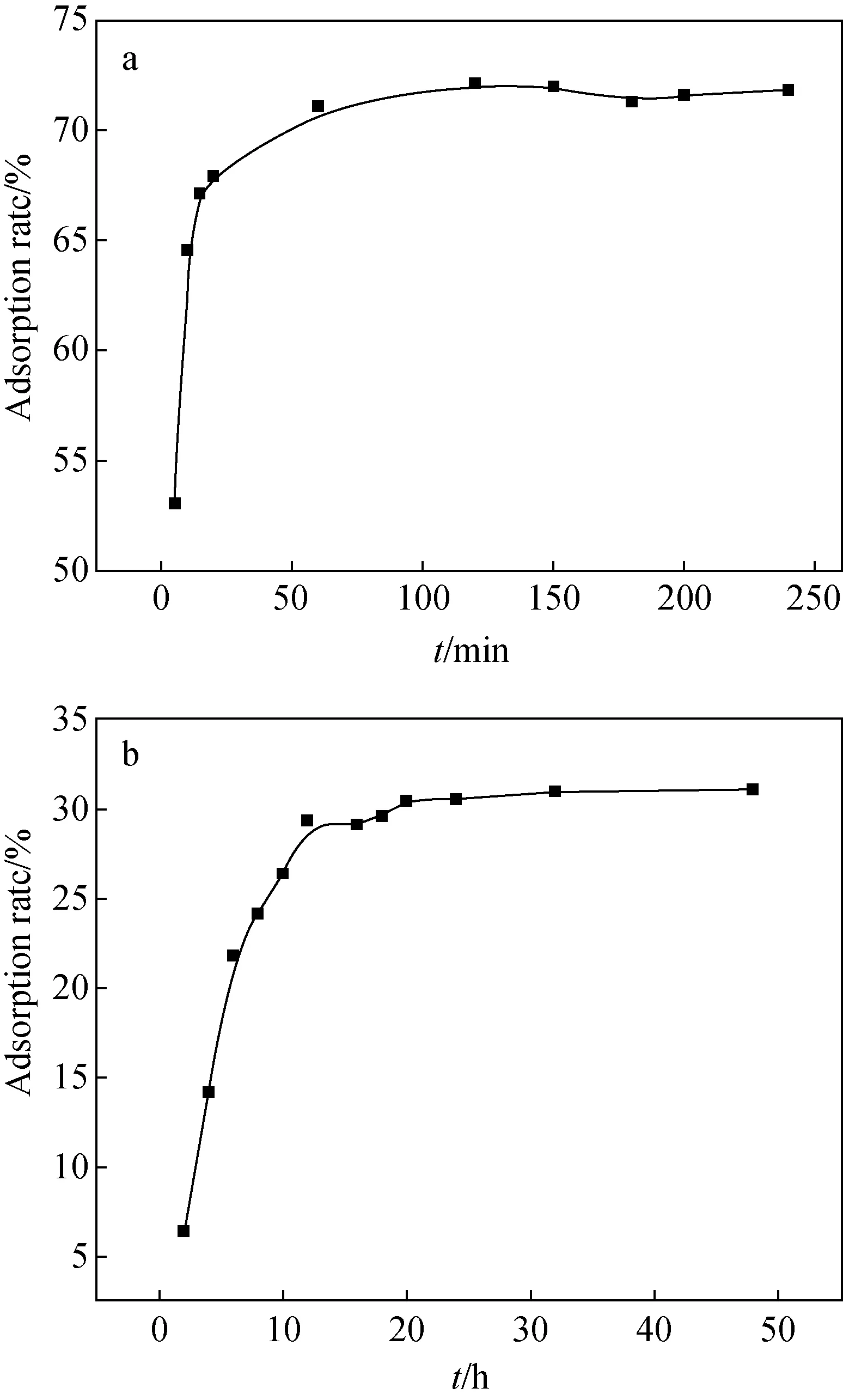
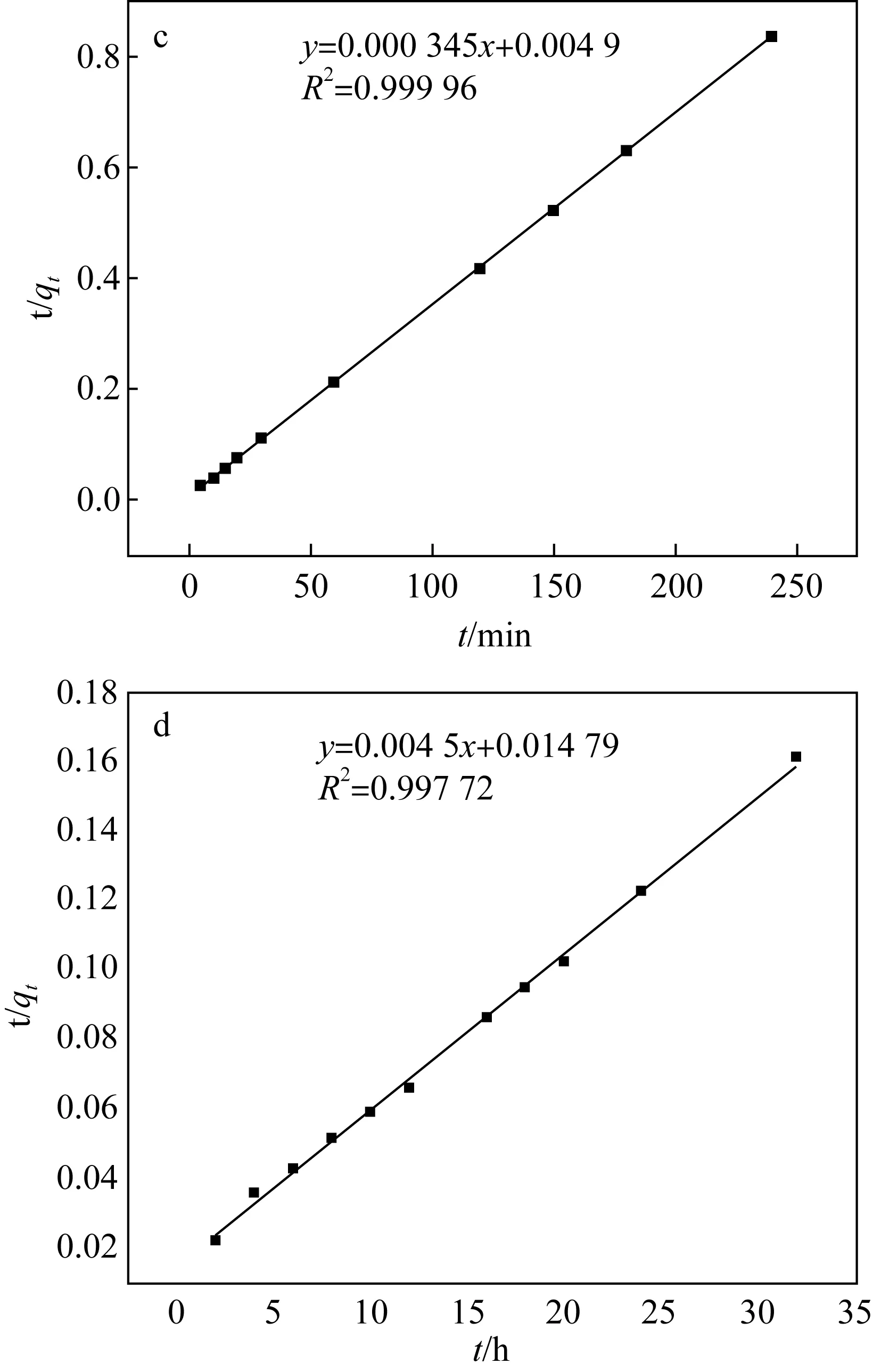
a、c Fe3O4@SiO2-NH2粒子上吸附時間對U(Ⅵ)吸附的影響和吸附的準二級動力學b、d 阿拉善黏土巖上吸附時間對U(Ⅵ)吸附的影響和吸附的準二級動力學圖4 吸附時間對U(Ⅵ)在不同吸附劑上的吸附影響及準二級動力學擬合Fig.4 Adsorption time and quasi-second-order kinetics of U(Ⅵ) on different adsorbents
為了進一步研究吸附過程,實驗數據使用準二級動力學方程來描述,方程式[13]如下式所示。準二級動力學線性模型為:
(4)
式中:k2(g/(μg·min))——準二級動力學的吸附速率常數。
吸附的準二級動力學擬合結果如圖4(c)和圖5(d)所示。根據二階動力學模型計算的qe值與實驗值qe很接近。因此,該吸附能很好地符合二階模型,吸附機制涉及化學吸附。
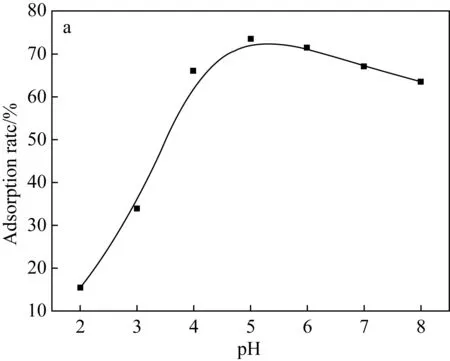
(a)Fe3O4@SiO2-NH2粒子上pH對吸附U(Ⅵ)的影響
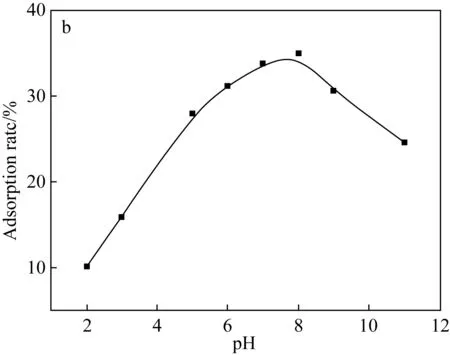
(b)阿拉善黏土巖上pH對吸附U(Ⅵ)的影響圖5 不同水相pH對U(Ⅵ)吸附的影響Fig.5 Effects of different aqueous pH on U(Ⅵ) adsorption
2.2.2 水相pH對U(Ⅵ)的吸附特征影響
由于地下水環境較為復雜,不同地區的地下水pH值有較大差異。因此,本文針對不同pH的地下水環境進行了實驗。pH調節至2~11,在室溫下,將固液混合物分別振蕩130 min和24 h,離心取上清液測量鈾濃度。測量結果如圖5所示,對于兩種吸吸附劑而言,在較酸性范圍內,對U(Ⅵ)的吸附率隨溶液pH的增加而迅速增加,到一定值后開始下降,復合粒子在pH為5時對U(Ⅵ)的最大吸附效率為73.49%,阿拉善黏土巖對U(Ⅵ)吸附的最佳pH為8。在低pH下,阿拉善水相中存在著Fe3+、Ca2+、Mg2+等大量陽離子以及H3O+占據了吸附劑表面上的大部分吸附位點,它們與UO22+形成競爭吸附。隨著pH增加至中性區域,主要通過減少H3O+的量來暴露吸附位點。U(Ⅵ)主要以UO22+的形式存在,吸附速率增加,這可能是由于U(Ⅵ)主要以(UO2)3(OH)5+的形式存在。隨著pH增加超過8,對U(Ⅵ)的吸附速率降低,首先這可能是因為UO22+離子水解形成不溶性氧化物4UO3·9 H2O和氫氧化物UO2(OH)2沉淀[14-16],使可溶性U(Ⅵ)含量下降;其次阿拉善水相中存在的陽離子容易在堿性條件下形成懸浮物對U(Ⅵ)的吸附起到抑制作用。
2.2.3 固液比對U(Ⅵ)在吸附劑上吸附特征的影響
室溫時,在各自最佳pH、吸附平衡時間和一定初始濃度的條件下,設計兩種吸附劑用量對U(Ⅵ)溶液進行吸附實驗,實驗結果如圖6所示。
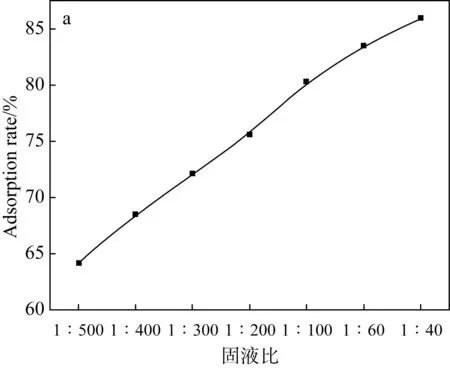
(a)Fe3O4@SiO2-NH2粒子上固液比對吸附U(Ⅵ)的影響
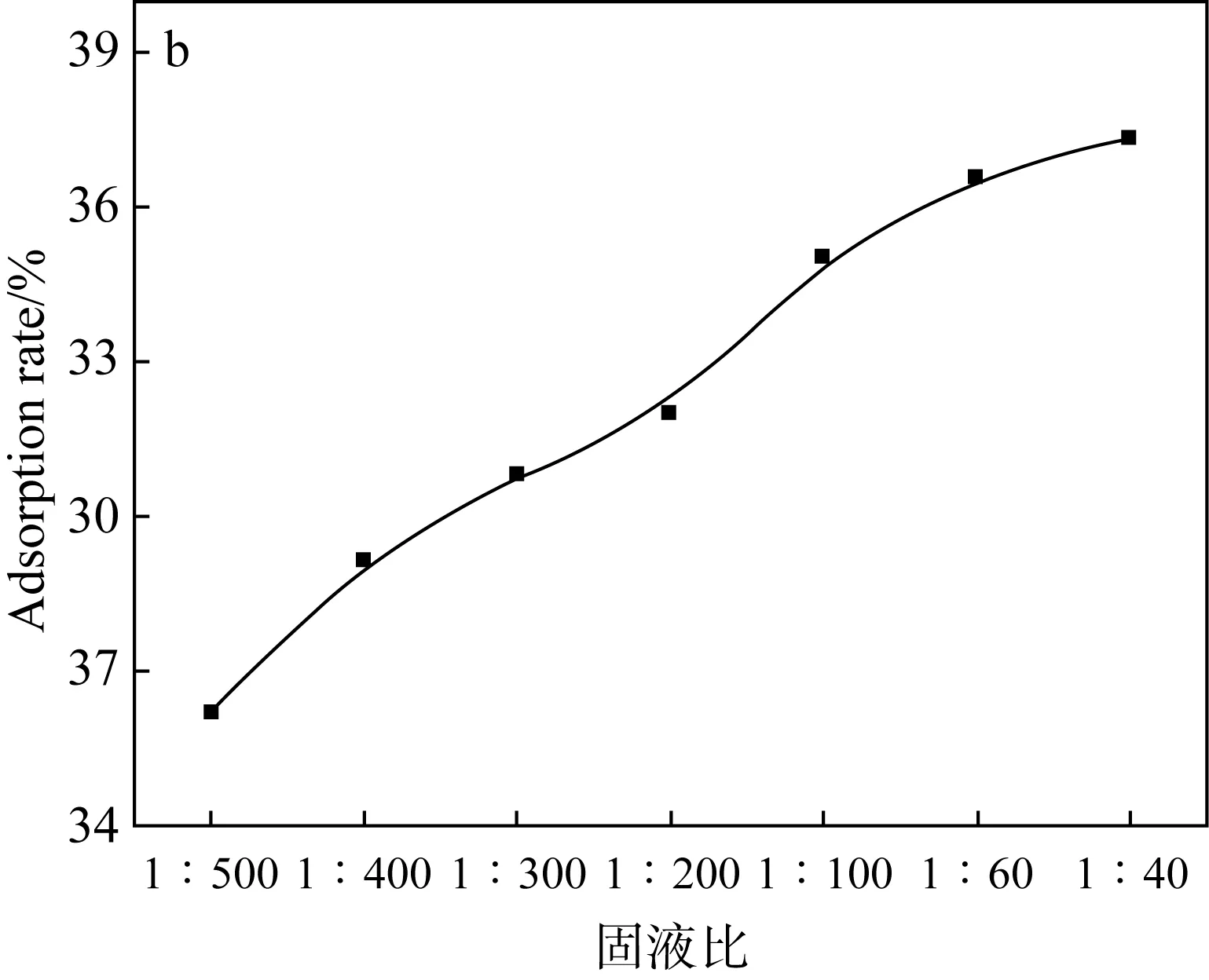
(b)阿拉善黏土巖上固液比對吸附U(Ⅵ)的影響圖6 不同固液比對U(Ⅵ)吸附的影響Fig.6 Effects of different solid-liquid ratios on U(Ⅵ)
吸附率隨著兩種吸附劑用量的增加而增大。這是由于吸附劑上活性位點的增加,因此使U(Ⅵ)更容易滲透到吸附劑中。在吸附劑用量達到一定值時,其吸附率趨向一個平緩的階段,可能是由于分離到單位質量吸附劑上的U(Ⅵ)減小引起的。
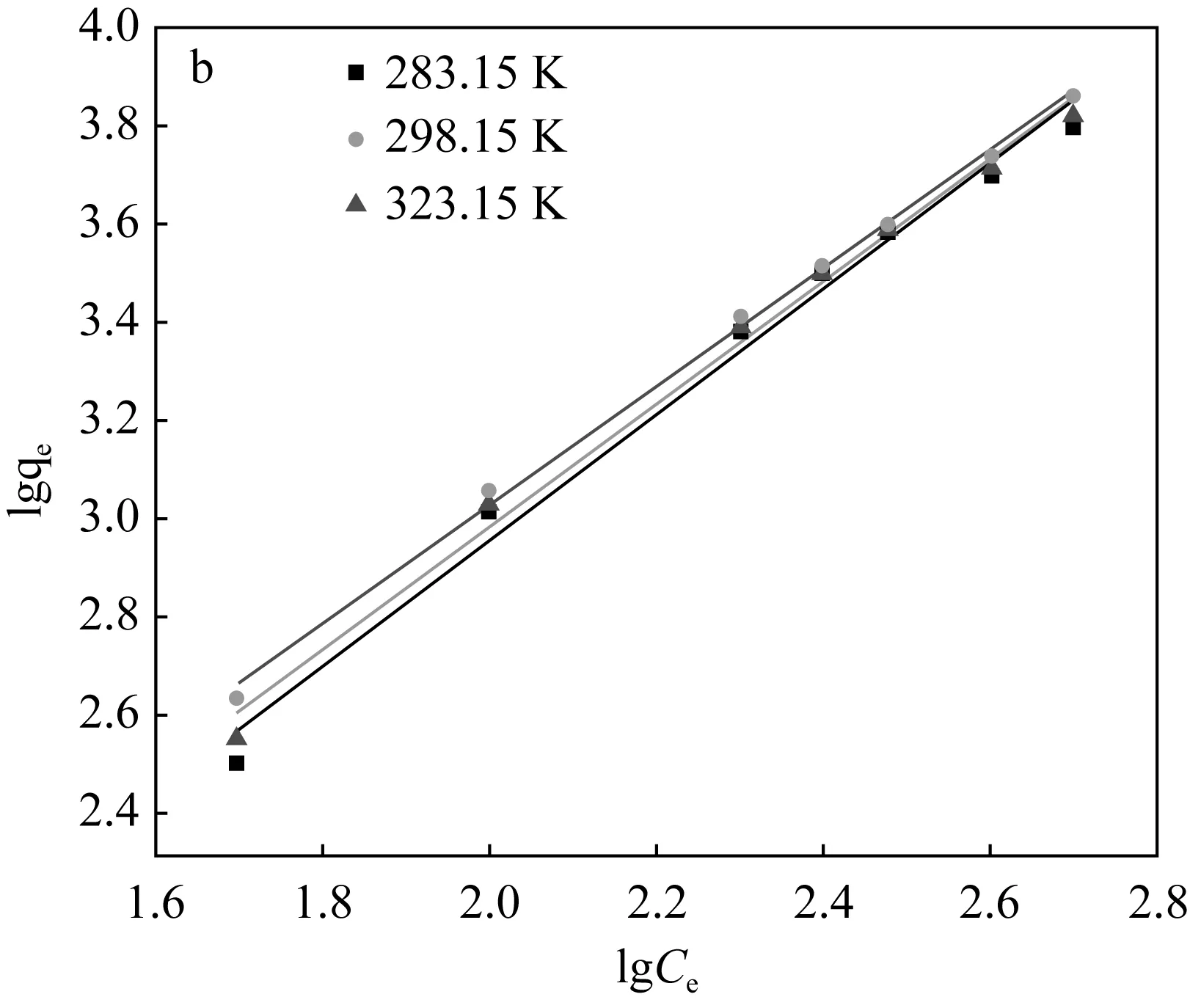
2.2.4 吸附等溫線
分別在pH為5和8的條件下,在283.15 K、298.15 K、323.15 K的溫度下設置U(Ⅵ) 的初始濃度為50~500 μg/mL進行吸附試驗,達到各自吸附平衡時間之后測其溶液中U(Ⅵ)的濃度。本實驗選用Freundich等溫模型對數據進行擬合,其相應模型對應的線性方程[17、18]分別為:
Freundich等溫模型:
(5)
式中:KL——Langmuir 吸附常數;
KF、n——Freundlich 吸附常數。
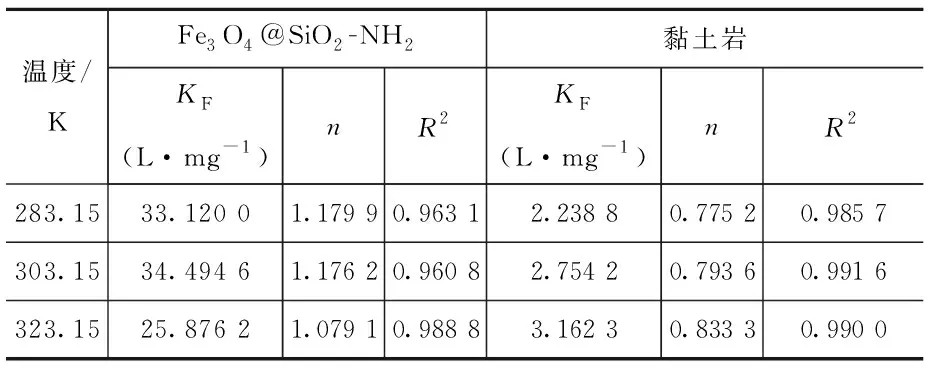
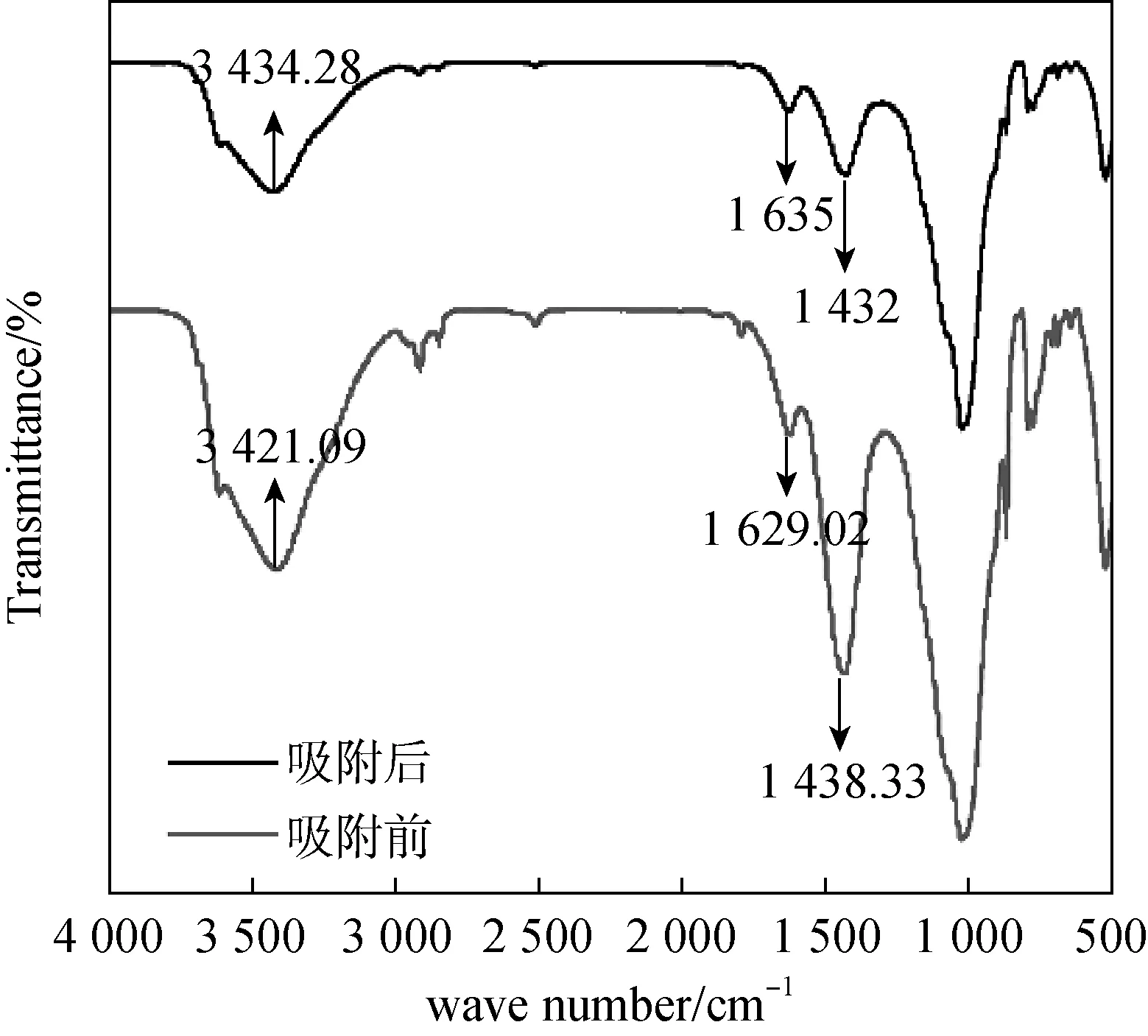
由圖7和表2可知,兩種吸附劑對U(Ⅵ)的吸附過程可以較好的用Freundlich吸附等溫模型進行模擬。說明該過程為多層吸附,且吸附質吸附后處于一個相對穩定的狀態,不會發生擴散或遷移。對于圖7(b)阿拉善黏土巖,0 (a)Fe3O4@SiO2-NH2粒子上吸附U(Ⅵ)的Freundlich等溫線 (b)阿拉善粘土巖上吸附U(Ⅵ)的Freundlich等溫線圖7 吸附劑對U(Ⅵ)的Freundlich吸附等溫線Fig.7 Freundlich adsorption isotherm of the adsorption of U(Ⅵ) on particles at different temperatures 表2 不同吸附劑對U(Ⅵ)吸附的Freundlich方程參數 圖8為黏土巖吸附U(Ⅵ)前后的對比譜圖,由圖譜內容可以看出,在U(Ⅵ)被吸附后黏土巖的峰形總體變化不大,且沒有新的譜帶出現,僅出現部分峰位的偏移,這說明黏土巖自身結構穩定,在對U(Ⅵ)發揮一定的吸附作用后不會引起過大的變化。在3 421 cm-1處的-OH峰向左發生了移動,同時峰強減弱; 1 629.02 cm-1處顯示為C=O的伸縮振動峰,吸附后的譜帶向左偏移了6 cm-1,且峰強增加;在1 438 cm-1處的C-O-H鍵伸縮振動峰向右發生了偏移,峰的強度也明顯增加[20-22]。 圖8 黏土巖吸附U(Ⅵ)前后的紅外光譜圖Fig.8 Infrared spectra of clay samples before and after U(Ⅵ) adsorption (1)SEM、TEM和FT-IR表征分析表明,成功制備出了Fe3O4@SiO2-NH2磁性復合粒子,粒徑均勻,分散良好,粒子呈微球狀,SiO2包裹于裸Fe3O4顆粒外表面,并有豐富的-NH2接枝。 (2)復合磁性粒子在阿拉善水溶液中對U(Ⅵ)的最佳吸附條件為:吸附溫度35 ℃,pH為5,吸附劑用量為0.04 g,平衡時間2 h,此時對U(Ⅵ)的平衡吸附率為73%;而阿拉善黏土巖對U(Ⅵ)的吸附率僅達35%左右,說明該復合粒子在阿拉善水相環境中比黏土巖具有更優良的吸附U(Ⅵ)的性能。 (3)阿拉善地下水復雜的組分以及酸度是影響U(Ⅵ)被去除的重要因素。兩種吸附劑對U(Ⅵ)的吸附過程中,準二級動力學方程可準確描述吸附過程,且能夠很好地符合Freundich等溫模型。整個吸附過程是通過吸附劑與吸附質之間的電子共用或靜電吸附來實現的,因此屬于化學吸附。同時實驗獲取的參數為U(Ⅵ)在特定吸附劑上的吸附模型的建立提供了重要依據,為推測核素鈾在環境中的遷移奠定了研究基礎。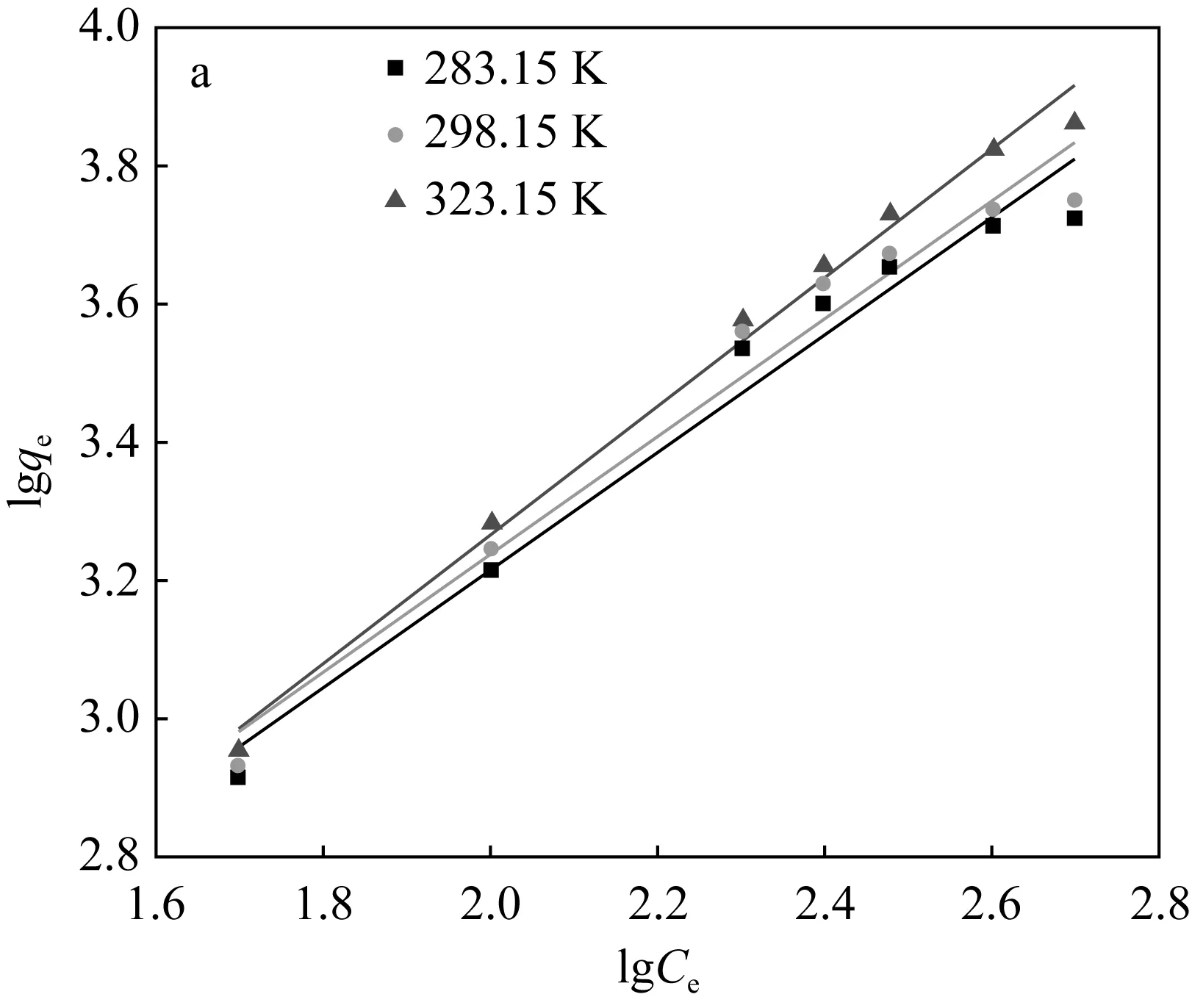
2.3 吸附前后紅外分析
3 結論
