雙硫侖及其代謝產物的抗感染藥理活性研究進展
2020-11-20 16:22:10游雪甫楊信怡
中國感染與化療雜志 2020年6期
孫 宇, 游雪甫, 楊信怡
雙硫侖(disulfiram)是二乙基二硫代氨基甲酸酯(diethyldithiocarbamate,DDC)的二聚體,起初通過臨床試驗證明雙硫侖通過抑制醛脫氫酶,阻礙乙醇在體內的代謝,致使飲酒者出現乙醛中毒樣癥狀,并將這種癥狀表現命名為“雙硫侖樣反應”。基于這一作用,雙硫侖被用作乙醇增敏藥,促使嗜酒者對乙醇產生厭惡感,從而用于戒酒治療[1-2]。當前,雙硫侖作為戒酒藥已被包括美國藥典、日本藥典、歐洲藥典在內的多國藥典收錄。雙硫侖安全性良好,口服用藥量可高達500 mg/d[3]。雙硫侖用于臨床后,其代謝產物DDC通過調節抗氧化酶活性對抗體內過多自由基而產生的抗白內障作用也被發現,并被認為是頗具潛力的抗白內障藥物[4]。近年來,研究還發現雙硫侖可通過抑制腫瘤干細胞生物標志物醛脫氫酶的作用抑制腫瘤干細胞以及調節NF-κB等信號傳導通路發揮抗腫瘤作用,其抗腫瘤機制的有效闡明[5-6],再次引起人們對該藥的廣泛關注。鑒于雙硫侖曾長期作為除螨劑使用[7],因此很多研究者開始關注其在抗微生物感染領域的藥用價值,進而關注其代謝產物的活性。本文針對雙硫侖及其主要代謝產物在抗感染研究領域的最新研究進展進行綜述,為相關領域研究者提供參考。

圖1 雙硫侖化學結構式
1 雙硫侖的藥動學特性
雙硫侖口服后,經胃腸黏膜吸收入血,在內源性硫醇和紅細胞中谷胱甘肽還原酶系統作用下迅速還原為單體DDC。后者并不穩定,可進一步降解為二乙胺(DEA)和二硫化碳(CS2)。同時,DDC也是Ⅱ相代謝酶系統的底物,在S-甲基轉移酶等Ⅱ相代謝酶催化下形成二乙基二硫代氨基甲酸甲酯(Me-DDC)和葡萄糖醛酸化的DDC。Me-DDC再經氧化脫硫轉化為二乙基硫代氨基甲酸甲酯(Me-DTC),Me-DTC則可被進一步氧化成其對應的砜和亞砜代謝物。吸收后,雙硫侖及其代謝產物均勻分布于全身各種組織中。口服或腹腔給予大鼠或小鼠35S-雙硫侖,在整個胃腸道以及血液、肝臟、腎臟、心臟、腎上腺、甲狀腺、胰腺、睪丸、脾臟、骨髓和肌肉中均可檢測到雙硫侖和DDC,而在紅細胞中未檢測到放射性。然而每種代謝物在各器官中的分布差異很大,這可能與不同組織中具有的生物轉化酶類型有關。雙硫侖和DDC在體外和體內均可與不同蛋白質的游離巰基結合形成復合二硫化物,在血液中,雙硫侖和Me-DTC主要與白蛋白高度結合。雙硫侖的代謝終產物主要通過腎、糞便和肺排出。雙硫侖口服后,約20.0%以原藥形式經糞便排出,大多數代謝產物包括DDC的葡萄糖醛酸苷或有機硫酸鹽通過腎臟消除,而CS2主要經肺排出[8]。
Faiman等[9]利用高效液相色譜(HPLC)技術在15名男性健康酗酒者中開展研究,獲得了雙硫侖單次和多次給藥后藥動學特性的詳細報告。在受試者服藥前和口服250 mg雙硫侖片劑后,在不同時間點分別收集其血液、尿液和呼吸樣本,獲得單次給藥樣本。72 h后,受試者需每天口服250 mg雙硫侖至第15天以獲得多次給藥樣本。受試者服用單劑量或多劑量的雙硫侖后,雙硫侖及其代謝產物的藥動學參數見表1。雙硫侖、DDC、Me-DDC、DEA、CS2的消除半衰期分別為7.3 h、15.5 h、22.1 h、13.9 h、8.9 h。給藥劑量為250 mg時,雙硫侖及其代謝產物平均達峰時間為8~10 h。但是雙硫侖及其代謝產物的血漿濃度存在明顯的個體間差異,這種差異可能由于雙硫侖的強脂溶性及其在脂肪組織中的分布、與血漿蛋白的結合力各不相同導致。藥物一旦在脂肪組織達到“飽和”,將產生趨于一致的血漿藥物水平。在對雙硫侖及其代謝產物DDC的細胞和實驗動物毒性研究中,未發現致癌、致畸或致突變作用[10]。
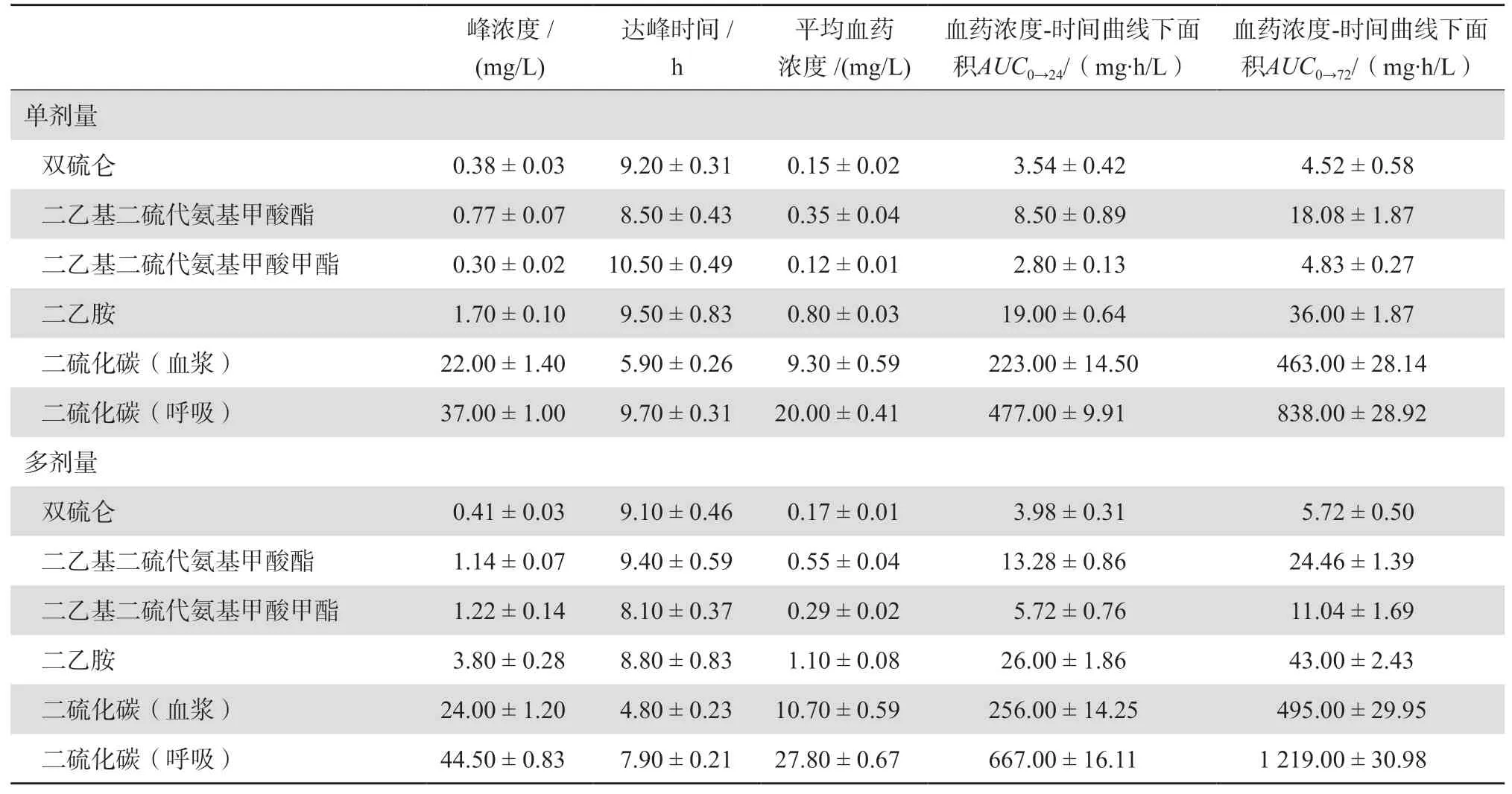
表1 雙硫侖及其代謝產物的藥動學參數
2 雙硫侖及其主要代謝產物的抗感染藥理活性
2.1 抗細菌活性
Frazier等[11]測定了雙硫侖及其主要代謝產物對30種革蘭陽性和陰性菌的抗菌活性。發現雙硫侖對除單核細胞增生李斯特菌和鏈球菌之外的大多數革蘭陽性菌均表現出抑菌活性。其中葡萄球菌(包括萬古霉素不敏感菌株)對雙硫侖的敏感性較高,最低抑菌濃度(MIC)為1~16 mg/L,而炭疽桿菌對雙硫侖及代謝產物DDC均表現出很高敏感性,MIC為0.5~4 mg/L,但其余幾種代謝產物未表現出明顯的抗菌活性。見表2。
耐甲氧西林金黃色葡萄球菌(MRSA)是引發臨床各類感染的重要病原,由于其耐多藥特性,往往致使病情遷延或抗感染治療失敗。Phillips等[12]發現體外條件下,雙硫侖對MRSA顯示較強的抑菌活性,MIC為1.33 mg/L。之后的研究發現雙硫侖除對耐多藥MRSA有較好體外抑菌活性,與萬古霉素聯用還對萬古霉素耐藥金黃色葡萄球菌(VRSA)具有協同抗菌效應,在研究中測得雙硫侖對臨床分離的MRSA和萬古霉素耐藥腸球菌(VRE)的MIC范圍為2~16 mg/L,對包括萬古
霉素中介金黃色葡萄球菌(VISA)和VRSA菌株在內的30株受試MRSA菌株,雙硫侖抑制90%菌株生長的MIC(MIC90)為16 mg/L,但革蘭陰性菌對雙硫侖敏感性低(MIC≥32 mg/L)[13]。棋盤法檢測萬古霉素和雙硫侖聯用,對萬古霉素A型VRSA菌株具有明顯的協同抗菌作用,對3株臨床分離VRSA菌株,雙硫侖(0.5~1 mg/L)將萬古霉素的MIC從>128 mg/L降至4~16 mg/L,抑菌濃度(FIC)指數 <0.5。這些研究提示雙硫侖或可作為萬古霉素增效劑或聯用藥物,用于糖肽類藥物治療無效的慢性和復發性MRSA感染,其在體內的聯用效果值得進一步關注。
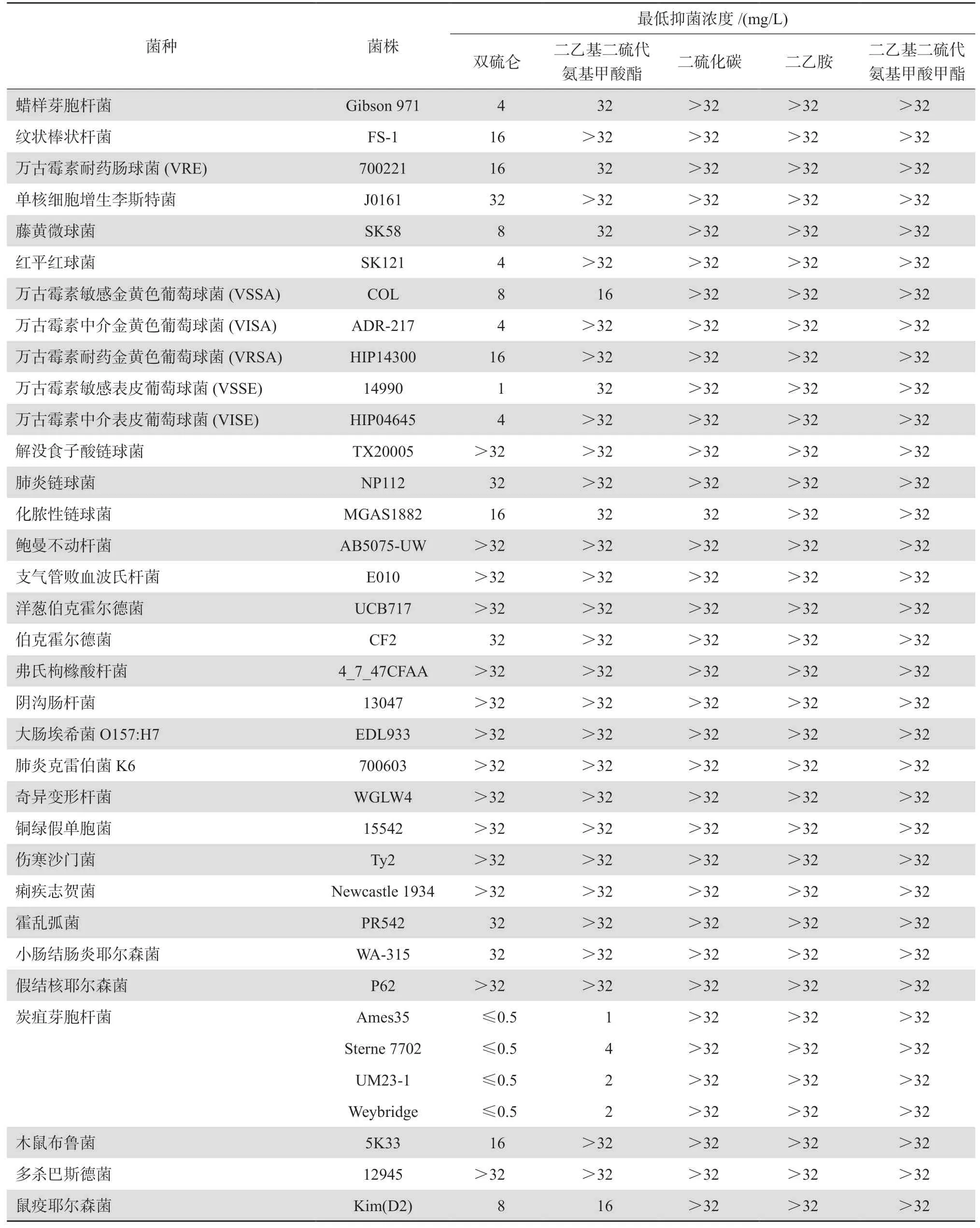
表2 雙硫侖及其代謝產物的抗菌活性
通過檢測雙硫侖及代謝產物在動物體內的分布,證實雙硫侖和DDC優先分布于肺部,這為雙硫侖治療某些感染性肺病如肺結核提供了依據[14]。早先,Jeney等[15]首次報道了雙硫侖和DDC在豚鼠結核分枝桿菌(Mycobacterium tuberculosis,Mtb)感染模型中的抗菌活性。Hubner等[16]進一步觀察到DDC在體外和體內條件下均可增強單核細胞的抗結核活性。在評價雙硫侖和DDC對耐多藥Mtb(MDR-Mtb)和泛耐藥Mtb(XDR-Mtb)菌株的抗菌活性中,對包括MDRMtb和XDR-Mtb菌株在內的42株臨床分離Mtb菌株,雙硫侖和DDC均顯示良好抗Mtb活性,MIC90分別為1.56 mg/L和3.13 mg/L,且與喹諾酮類藥物(左氧氟沙星、司帕沙星、環丙沙星)無交叉耐藥性[17]。隨后,研究者利用人單核細胞系THP-1檢測雙硫侖和DDC細胞內殺Mtb的活性,發現二者以劑量依賴方式分別在6~30 mg/ L和10~30 mg/ L對細胞內Mtb呈現明顯殺菌活性。在慢性結核病(TB)小鼠模型中,雙硫侖亦顯示較明顯的體內殺菌活性。雙硫侖治療組在80~160 mg/kg劑量時,小鼠肺和脾中菌落形成單位計數值(CFU)從105CFU/臟器降低至104CFU/臟器。盡管目前雙硫侖抗Mtb的作用機制仍不清楚,但有研究顯示雙硫侖結構類似物二硫代氨基甲酸鹽類可能通過抑制β碳酸酐酶(β-CAs)活性抑制Mtb的生長,該作用機制對于尋找雙硫侖及DDC的抗Mtb靶標具有一定啟示意義。總之,雙硫侖及其代謝物DDC可能是一類潛在的治療MDR / XDR-TB的新型藥物,其抗Mtb作用機制值得進一步關注。
2.2 抗真菌活性
免疫功能低下患者如艾滋病、癌癥晚期、重癥監護患者易發生真菌感染,目前常用的抗真菌藥往往因真菌的跨膜外排機制[如P-糖蛋白(P-gp)介導的藥物外排]而活性降低[18]。研究顯示,雙硫侖對真菌細胞膜表面的P-gp有抑制調節作用。Sauna等[19]對雙硫侖作為多藥耐藥調節劑的分子基礎進行探索,發現雙硫侖能與多種ATP結合轉運蛋白的藥物-底物結合位點結合并抑制耐藥性相關的ATP水解。以抗深部真菌的三唑類藥物為例,真菌對其產生耐藥性主要由P-gp介導。而雙硫侖作為一種外排泵調節劑,可通過抑制這種藥物外排克服真菌對三唑類藥物(主要是氟康唑耐藥白念珠菌)的耐藥性,從而可作為一種潛在的聯用藥物,輔助治療耐藥性真菌引起的感染。
受此啟發,Khan等[20]采用氟康唑聯合雙硫侖對白念珠菌和光滑念珠菌進行抗真菌測試,然而并未觀察到兩藥存在任何協同或相加作用,他們推測可能是由于14-α-脫甲基酶的突變所致。之后,他們又對雙硫侖的抗一系列酵母和絲狀真菌活性進行了檢測。研究顯示,雙硫侖對61株受試氟康唑敏感或耐藥的白念珠菌、非白念珠菌、絲狀真菌分離株均顯示良好殺真菌活性,MIC50、MIC90分別為4 mg/L、8 mg/L。雙硫侖對試驗中13株曲霉也呈現明顯的殺真菌活性,MIC50、MIC90分別為2 mg/L、8 mg/L。盡管目前人們已觀察到雙硫侖具有較顯著的殺真菌活性,但其作用機制仍待闡明。
2.3 抗寄生蟲活性
早在1942年,Gordon等[7]就提出四乙基結構類似物應具有殺螨作用,進而利用背肛螨屬螨蟲的感染大鼠模型研究了雙硫侖的殺螨作用,并與2種疥瘡治療藥物苯甲酸芐酯和二甲基噻吩(滅疥)進行了療效比較。結果顯示,雙硫侖的殺螨作用較2種對照藥更為高效、迅速,應是一種有效的疥瘡治療藥物。這一發現為雙硫侖用于疥瘡治療提供了依據,并啟發人們進一步探索其治療其他寄生蟲感染的可能。雙硫侖上市后,人們對其抗寄生蟲活性的研究繼續深入,后續研究顯示其對利什曼蟲、藍氏賈第鞭毛蟲(賈第蟲)、惡性瘧原蟲、克氏錐蟲、陰道毛滴蟲等具有廣譜殺蟲活性。
利什曼蟲感染可導致內臟或皮膚利什曼蟲病,且一旦引起內臟利什曼蟲病,如未及時接受治療往往危及生命。Osorio等[21]采用熒光素酶轉染的杜氏利什曼原蟲,對倉鼠的離體脾臟進行感染制成外植體培養系統,篩選抗蟲劑。最終從4 000多個化合物中篩得雙硫侖,發現其在納摩爾濃度具有顯著殺利什曼原蟲活性。Peniche等[22]通過進一步研究,發現雙硫侖與二價金屬鹽(1 μmol/ L)合用,能顯著提高殺滅利什曼原蟲活性。例如,雙硫侖與硫酸鋅組合可使針對杜氏利什曼原蟲的體外治療指數增加1.8倍。研究人員通過對利什曼蟲進行代謝網絡計算機模擬評估,預測雙硫侖的抗利什曼原蟲活性可能是與其干擾蟲體中由LmjF25.1170和LmjF25.1180基因編碼的跨膜質子轉運系統有關。這些基因編碼的蛋白質是F型ATP酶β鏈復合物組成部分,參與寄生蟲ATP合成,通過特異性跨膜轉運質子(H+)產生電化學梯度,從而為ATP合成提供動力。在杜氏利什曼蟲中,雙硫侖導致F-ATP合成酶的抑制使線粒體膜電位去極化,ATP合成受損,從而增加細胞活性氧的產生,導致寄生蟲DNA斷裂而死亡。盡管目前尚無單用或聯用雙硫侖治療皮膚或內臟利什曼病的臨床數據,但這些研究確為雙硫侖未來用于利什曼病的治療奠定了理論基礎。
賈第蟲是一種常致人體腸道感染的寄生蟲,可致賈第蟲病。Nash等[23]發現體外條件下測得(1.23±0.32)μmol/L雙硫侖可殺滅賈第蟲。在該寄生蟲感染的小鼠模型中,研究者進一步觀測到每天2次,持續4 d灌胃給予雙硫侖25 mg,可使40.0%小鼠治愈,未治愈小鼠的賈第蟲負荷也有顯著性降低(P<0.01),而對照組治愈率為0。Hill等[24]在體外、體內條件下觀測了雙硫侖對寄生性鼠鞭蟲卵殼形態的影響。研究人員在小鼠感染鼠鞭蟲后第25天開始每天分別給予劑量為2.5 mg/kg、5.0 mg/kg、7.5 mg/ kg的雙硫侖,治療26 d后,觀察到治療組所有成年雌性鞭毛蟲所產蟲卵均為畸形卵。此外,與對照組小鼠比較,5.0 mg·kg-1·d-1、7.5 mg·kg-1·d-1劑量的雙硫侖治療組小鼠尸檢中發現的成蟲數量顯著降低(P<0.05)。一般認為,鼠鞭蟲的卵殼可保護蟲卵在發育過程中免受外界傷害,在卵殼形成過程中,聯苯酚氧化酶發揮著重要作用。研究者推測,雙硫侖作為一種氧化抑制劑,可能抑制該酶活性進而干擾蟲卵孵化為成蟲。雙硫侖通過干擾卵殼形成,阻斷由卵發育為鞭蟲成蟲的殺蟲方式具有重要臨床意義。
此外,大量報道還顯示雙硫侖和/或其代謝產物在體外對惡性瘧原蟲、克氏錐蟲、陰道毛滴蟲也具有抑制活性。Scheibel等[25]最早注意到雙硫侖和DDC的抗瘧活性,他們認為這兩個化
合物具有的抑制多種金屬蛋白酶加氧酶的活性、良好的脂/水分配系數、與金屬離子的高結合常數等,均有利于針對瘧原蟲發揮選擇性毒性。體外研究結果顯示,雙硫侖和DDC在濃度僅為0.1 mg/L時即表現出明顯的抗瘧活性。在該研究基礎上,Meshnick等[26]還發現DDC可通過與外源性銅離子或內源性銅離子(存在于宿主細胞質和寄生蟲溶酶體中負載超氧化物歧化酶的紅細胞中)形成復合物,導致蟲體細胞膜損傷,進而發揮抗瘧作用。不過,DDC-Cu復合物誘導膜損傷的具體機制,目前仍不清楚。Lane等[27]在體外檢測了雙硫侖及其還原代謝產物二乙胺-N-二硫代碳酸鈉(DECD)對克氏錐蟲包括上鞭毛體、錐鞭毛體、無鞭毛體形式的殺滅活性。結果顯示,在50 mg/L濃度下,雙硫侖和DECD對上鞭毛體的抑制率分別為64.6%和69.7%,對組織培養錐鞭毛體的抑制率分別為 47.7%和46.1%,對無鞭毛體感染的3T3成纖維細胞抑制率分別為60%和67%。研究者同時指出雙硫侖和DECD殺錐蟲的潛在機制與干擾克氏錐蟲的必需金屬離子代謝有關,并認為二者均可作為預防或治療錐蟲病的藥物候選物。另外,雙硫侖和DDC對甲硝唑敏感及耐藥的陰道毛滴蟲和胎三毛滴蟲也具有明顯抑制活性,雙硫侖和DDC對甲硝唑敏感、耐藥陰道毛滴蟲的MIC分別為0.1~0.7 μmol/ L、0.2~1.3 μmol/ L和0.3~9 μmol/ L、1.2~9 μmol/L;雙硫侖和DDC對甲硝唑敏感、耐藥的胎三毛滴蟲的MIC分別為0.1~1.0 μmol/L、1.0~6.9 μmol/L和0.3~1.3 μmol/L、0.6~6 μmol/L[28]。總之,上述多項研究均顯示雙硫侖及其代謝產物在治療寄生蟲感染方面具有巨大應用潛力。
2.4 抗病毒活性
1975年,Cari?-Lazar等[29]報道了雙硫侖可影響包膜病毒的增殖。在病毒吸附前1 h和吸附期間加入雙硫侖(0.1~0.3 mmol/L),對塞姆利基森林病毒(semliki forest virus,SFV)、禽瘟病毒(fowl plague virus)、新城疫病毒(newcastle disease virus)、水皰性口炎病毒(vesicular stomatitis virus)、偽狂犬病病毒(pseudorabies virus)的增殖均有明顯抑制作用。不過,在病毒吸附至雞胚細胞后再加入藥物,則不能觀察到病毒增殖受抑的現象。雙硫侖既不干擾病毒的受體也不干擾紅細胞,并且不會阻止病毒吸附,故研究者認為雙硫侖可能影響病毒繁殖的早期步驟。
聯合抗反轉錄病毒療法(cART)是一種抑制人類免疫缺陷病毒(HIV)復制、延緩病情發展的有效治療方案,但這一療法無法根除病毒感染,主要是因為HIV的基因組可通過反轉錄過程穩定整合入宿主細胞如記憶性CD4+T細胞的染色體中,進入潛伏狀態,表現為長期的轉錄沉默[30]。針對此現象,當前的主要治療策略是重新激活潛伏的HIV,誘導受感染的細胞繼續產生病毒顆粒,從而使受感染的宿主細胞死亡或誘導HIV特異性T 細胞殺傷。Xing等[31]用抗凋亡蛋白Bcl-2轉染的原代CD4+T細胞模型高通量篩選能誘導病毒基因表達但不觸發細胞活化的化合物,發現雙硫侖可重新激活潛伏感染的HIV-1而不引起總體T細胞的活化。目前認為,雙硫侖通過消耗細胞內磷酸酶和張力蛋白同源體(PTEN)上調蛋白激酶B(Akt)信號通路,從而誘導HIV-1潛伏感染的單核細胞系中HIV-1基因轉錄[32]。在此基礎上,Spivak等[33]在臨床試驗中,連續14 d給予HIV-1患者雙硫侖500 mg/ d,觀察到該藥確實可以重新激活人體靜息記憶CD4+T細胞中潛伏的HIV-1。隨后,Elliott等[34]又進行了一項劑量遞增研究,他們對30例接受cART治療的AIDS患者,連續3 d每天分別給予500 mg、1 000 mg和2 000 mg劑量的雙硫侖,結果導致所有劑量組受試者均出現了細胞相關的未拼接 HIV RNA增加,且在高劑量下觀察到血漿HIV RNA的增加,與激活HIV潛伏期一致。該試驗同時發現,即使雙硫侖用藥量達到常規解酒用藥量的4倍,患者依然耐受良好。鑒于其良好的安全性表現,雙硫侖應該適用于長期、聯合給藥,用于cART中潛伏HIV的激活。
鑒于雙硫侖能與銅離子螯合,Levinson等[35]研究了雙硫侖及其代謝產物DDC對勞氏肉瘤病毒惡性轉化、真核細胞合成和核酸結合的影響。結果發現,雙硫侖可抑制勞氏肉瘤病毒RNA依賴性DNA聚合酶的活性,并使病毒喪失惡性轉化雞胚細胞的能力,但DDC對病毒無明顯影響。雙硫侖和DDC誘導正常雞胚和人類包皮細胞中四種蛋白質的合成,當銅離子存在時,DDC可與HeLa細胞DNA和勞氏肉瘤病毒70S基因組RNA結合。研究者認為,雙硫侖可能是通過與DNA聚合酶活性位點中的Zn2+結合而對病毒粒子DNA聚合酶產生抑制,從而抑制勞氏肉瘤病毒轉化活性。
病毒蛋白中,Zn2+是維持天然蛋白質結構穩定的關鍵輔助因子。某些與Zn2+結合的半胱氨酸可與親電試劑反應,致使Zn2+失去配體而被排出,從而導致天然蛋白質結構和功能受到破壞。故病毒的某些重要結構或功能蛋白中,這類含有反應性鋅結合半胱氨酸殘基的不穩定鋅位點可能成為一種頗具希望的抗反轉錄病毒治療靶標。雙硫侖作為一種經臨床證明、安全性頗高的鋅驅逐劑,顯然可通過靶向鋅位點而成為一種潛在的抗病毒藥物。Lee等[36]在丙型肝炎病毒(HCV)的NS5A蛋白中鑒定出不穩定的鋅位點,并證明雙硫侖可以從預測的靶位將Zn2+驅除,從而產生抑制HCV復制的活性,且抑制程度與抗病毒藥物利巴韋林相似,由此認為,雙硫侖與干擾素和/或直接靶向HCV的抗病毒藥物聯用可以治療感染HCV并伴有酒精依賴的患者。Novo-Veleiro等[37]也指出,由于飲酒和HCV感染具有協同肝毒性作用,兩種因素共存可增加晚期肝硬化和患肝癌的風險,因此對存在酒精依賴的HCV感染患者采用雙硫侖治療,可能會對抗病毒療效和患者依從性產生積極影響,為治療提供額外支持。此外,Chen等[38]通過RNA干擾基因組文庫篩選,發現ZBTB25是多種細胞基因的轉錄抑制因子,涉及ZBTB25的蛋白質-蛋白質和蛋白質-RNA相互作用促進病毒RNA轉錄和復制,而ZBTB25相關功能需要完整的鋅指結構域和ZBTB25的翻譯后SUMO-1修飾。研究者發現用雙硫侖處理過表達ZBTB25的細胞,由于RNA合成減少導致甲型流感病毒的產生顯著降低,雙硫侖破壞鋅指結構的功能有效抑制了甲型流感病毒復 制。
另外,對于具有高致病性的嚴重急性呼吸綜合征冠狀病毒(SARS-CoV)和中東呼吸綜合征冠狀病毒(MERS-CoV),雙硫侖對兩者的木瓜蛋白酶樣蛋白酶(PLpros)分別產生競爭性抑制和別構抑制作用。除去未結合的雙硫侖后,病毒出現的緩慢結合抑制現象和酶活性的不可恢復性,表明雙硫侖很可能通過共價結合滅活了SARS-CoV PLpro,而雙硫侖和6-巰基鳥嘌呤或霉酚酸聯用對MERS-CoV PLpro顯示出的協同抑制作用則提示這3種臨床藥物聯合使用,可能具有治療冠狀病毒感染的潛在價值[39]。
3 結語
雙硫侖藥用歷史悠久,生物利用度高(80.0%),安全性良好。在當今藥物研發領域,作為“老藥新用”策略中的一個代表,雙硫侖及部分代謝產物表現出廣泛的藥理學活性,具有極高的藥用研究價值和發掘潛力。在抗感染治療研究領域,雙硫侖單用或與其他藥物聯用,在抑制耐藥金黃色葡萄球菌、Mtb生長,殺滅多種真菌、寄生蟲,抑制病毒轉錄、復制、激化等方面展現出良好活性,為其作為抗感染藥物的進一步開發提供了可能。不過,與雙硫侖抗感染相關的許多作用機制仍不明確,因此還需基于更多藥理研究模型,提出合理科學假說,深入探索其有效作用靶標,并進行可能的結構改造,為其成為一類新型抗感染藥物提供依據。
