miR-17在體外腦缺血損傷中對Smad錨著蛋白的負性調控作用
2020-12-25 02:45:46石曉花王姣琦徐忠信
中國實驗診斷學 2020年12期
石曉花,董 玥,付 超,徐 磊,袁 罡,莽 靖,王姣琦*,徐忠信*
(1.吉林大學中日聯誼醫院 神經內科,吉林 長春130033;2.吉林大學中日聯誼醫院 神經外科,吉林 長春130033)
激活素A(Activin A,ActA)是目前較為公認的神經保護因子[1]。研究發現,在體內外缺血性腦損傷中,ActA主要通過下游Smads信號通路發揮神經保護、抗氧化應激和自噬調控等作用[2,3]。課題組近期研究發現,缺血性腦損傷后ActA/Smads信號僅短時活化,隨后在Smad錨捉蛋白(Smad anchor for receptor activation,SARA)的調控下出現自發衰減[4,5]。研究發現,microRNA-17(miR-17)可通過調控SARA的表達阻斷食管鱗狀細胞癌的遷移和侵襲[6]。在腦缺血損傷過程中,miR-17是否通過SARA調控ActA/Smads信號尚不清楚。為探討該問題,本研究通過miR-17 mimic轉染及雙熒光素酶報告基因檢測,初步探討了miR-17在體外腦缺血損傷過程中對SARA的調控作用,為延長ActA/Smads信號活化時程,提高ActA神經保護作用提供潛在干預位點。
1 材料與方法
1.1 主要材料
大鼠高分化腎上腺嗜鉻細胞瘤(PC12 細胞)購自上海生物科學院細胞資源中心。miRNA qRT-PCR試劑盒、riboFECTTM CP Reagent轉染試劑、雙熒光素酶報告基因載體、mimic及陰性對照NC購自廣州銳博公司。兔抗大鼠SARA抗體購自美國Epitomics公司;二抗購自北京博奧森生物技術有限公司。
1.2 實驗方法
1.2.1細胞的培養及OGD模型的復制 高分化PC12細胞在10 %胎牛血清的高糖DMEM中培養,在37℃,5 % CO2的潮濕培養箱中孵育。利用含連二亞硫酸鈉(NaS2O4,終濃度1 mM)的無糖DMEM培養液在37℃,5% CO2、95% N2的培養箱中氧糖剝奪(Oxygen Glucose Deprivation,OGD)處理細胞[5]。OGD計時0、1.5、3、6、12 h。
1.2.2miR-17的表達檢測 為檢測miR-17在體外氧糖剝奪模型中的表達變化,本研究收集OGD不同處理時長的高分化PC12細胞,Trizol法提取總RNA,使用miR-qRT-PCR試劑盒,按照說明書步驟進行加尾、逆轉錄和實時定量PCR檢測,以U6為內參對照。基因引物如下:miR-17逆轉錄引物:5′-CTCAACTGGTGTCGTGGAGTCG GCAATTCAGTTGAGCTACCTGC-3′,上游引物:5′-ACACTCCAGCTGGGC AAAGTGCTTACAGTGC-3′,下游引物:5′-TGGTGTCGTGGAGTCG-3′;U6 逆轉錄引物:5′-AACGCTTCACGAATTTGCGT-3′,上游引物:5′-CTCGC TTCGGCAGCACA-3′,下游引物:5′-AACGCTTCACGAATTTGCGT-3′。應用ABI7500Fast 型實時熒光定量PCR儀進行熒光收集及分析。PCR運行參數:95℃預變性20 s,然后95℃變性10 s,60℃退火20 s,70℃延伸10 s,共40個循環。應用Relative Quantification (ddCt) Study進行相對表達量(relative quantity,RQ)RQ分析,計算SARA基因的相對表達水平。檢測實驗重復3次。
1.2.3miR-17 mimic轉染 將生長狀態良好的高分化PC12細胞接種于6孔板,細胞密度達70-80%。次日更換6孔板內的細胞培養液為無血清的高糖DMEM,按照說明書步驟進行轉染操作。轉染mimic的終濃度為50 nM。轉染6 h后將培養液更換為含血清的完全培養液。在轉染24 h、48 h和72 h分別收集細胞,每組3個復孔。
1.2.4Western blot蛋白水平表達檢測 將轉染不同時間分組的細胞提取總蛋白,BCA法測定蛋白濃度,調整上樣量。按照操作步驟進行電泳、轉膜和封閉,使用含5 %脫脂奶粉的TBST溶液稀釋一抗(1∶1000),4 ℃孵育過夜后洗膜,二抗(1∶1000)室溫孵育1 h,洗膜后ECL顯影壓膠片。凝膠圖像分析,掃描膠片,吸光度分析,計算目的條帶與相應內參條帶的灰度值,使用兩者的比值表示目的蛋白的表達水平。
1.2.5雙熒光素酶報告基因檢測 SARA又稱Zfyve9。利用MiRDB(http://mirdb.org/)和TargetScan(http://www.targetscan.org/)在線分析軟件,預測miR-17對SARA 3’ UTR區的潛在調控位點:GCACTTTA(57-64)和GCACTTT(403-409)。將SARA 3’ UTR區克隆至pmiR-RB-REPORTTM雙熒光素酶報告載體。根據兩個預測位點,委托廣州銳博公司設計并合成野生型及突變型報告載體,分別為:R-Zfyve9-WT1(野生型)、R-Zfyve9-MUT1(57-64位突變,CGTGAAAT)、R-Zfyve9-MUT2(403-409位突變,CGTGAAA)和R-Zfyve9-MUT3(57-64位突變,CGTGAAAT和403-409位突變,CGTGAAA)。所有載體的報告熒光為位于目的基因3’ UTR區上游的海腎熒光素酶基因(hRluc),校正熒光為螢光蟲熒光素酶基因(hluc)。將miR-17 mimic(終濃度50 nM)與報告基因載體(250 ng/孔)使用LipofectamineTM2000共轉染接種于96孔板的293T細胞,每組3復孔。轉染6 h后更換新鮮培養液。轉染48 h后吸出培養基,加入luciferase底物,振蕩10 min后加入Stop reagent,振蕩后熒光照度計測定熒光值。通過報告基因相對熒光值的下調來印證miRNA與靶基因的相互作用。
1.3 統計學分析
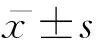
2 結果
2.1 miR-17在體外缺血損傷中的動態變化
利用實時定量PCR技術檢測miR-17的表達情況。結果miR-17在OGD早期1.5 h即可檢測到表達上調,至OGD 3 h時達到高峰,超過OGD 0 h組10倍(P<0.05,如圖1)。隨著OGD時間延長,miR-17表達顯著降低,到OGD 6 h和12 h時表達量與OGD 0 h比較差別無統計學意義(P>0.05)。
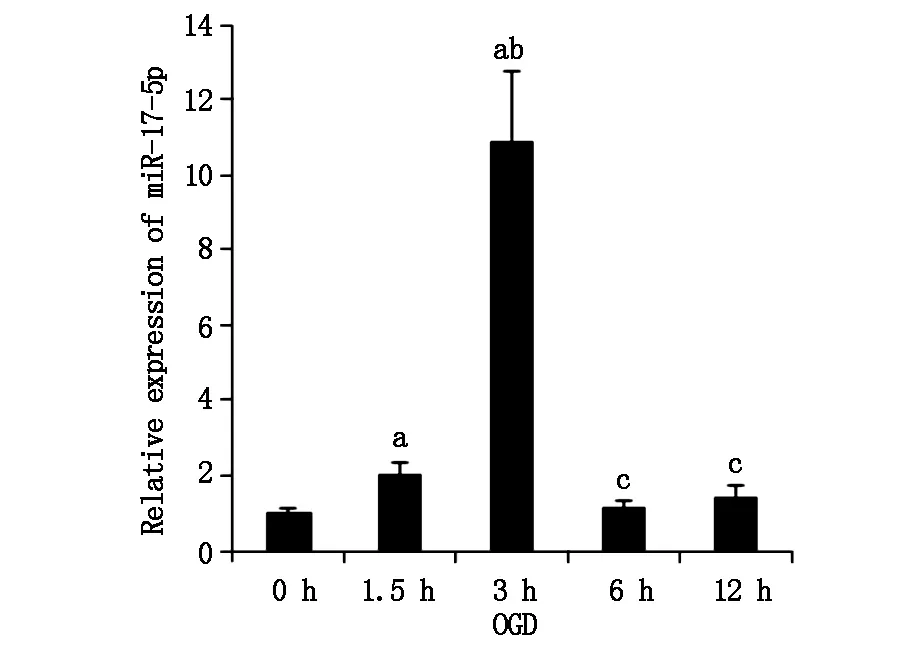
圖1 miR-17表達隨OGD動態變化a與OGD 0 h組比較P<0.05,b與OGD 1.5 h組比較P<0.05,c與OGD 3 h組比較P<0.05,n≥3/組
2.2 miR-17下調SARA蛋白表達
為明確miR-17對SARA的表達調控作用,本研究利用miR-17 mimic轉染高分化PC12細胞,并使用Western blot技術對轉染不同時間的SARA蛋白水平進行檢測。結果,不同轉染時間的mimic NC組SARA蛋白表達水平與對照組(control)相比,差別無統計學意義(P>0.05)。miR-17 mimic轉染抑制SARA蛋白表達,轉染24 h組SARA蛋白水平下調了57.01%。隨著轉染時間的延長,miR-17 mimic轉染組SARA蛋白水平雖略有回升,但miR-17 mimic仍能有效抑制SARA蛋白表達直至mimic轉染72 h(P<0.05,如圖2)。
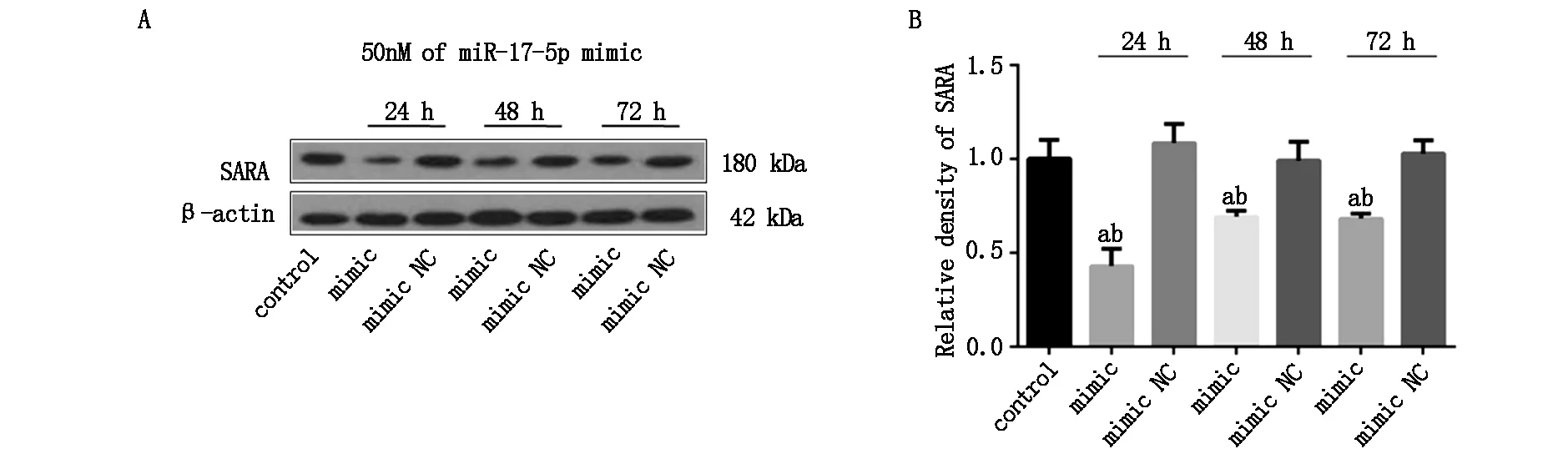
圖2 miR-17 mimic抑制SARA蛋白表達(A)SARA蛋白表達檢測;(B)SARA蛋白表達的灰度分析(a與對照組比較P<0.05,b與相應轉染時間的mimic NC組比較P<0.05,n≥3/組)
2.3 miR-17靶向調控SARA表達
R-Zfyve9-WT1、R-Zfyve9-MUT1、R-Zfyve9-MUT2和R-Zfyve9-MUT3 4種雙熒光素酶報告基因載體分別與miR-17 mimic或 NC轉染后熒光分析示: 與R-Zfyve9-WT1+NC組比較,R-Zfyve9-WT1+miR-17 mimic組報告熒光顯著下調,說明miR-17對SARA基因的3’UTR 區存在靶向調控。對兩個預測靶位點進行突變后發現,突變型載體Mut1、Mut2、Mut3與miR-17 mimic共轉染熒光強度較R-Zfyve9-WT1+miR-17 mimic組明顯恢復(P<0.05,如圖3)。說明位點(57-64)和位點(403-409)是miR-17調控SARA表達的靶點,且位點(57-64)和位點(403-409)的調控功能上呈現一定的協同作用。
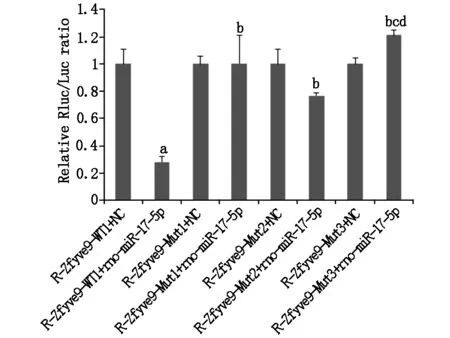
圖3 雙熒光素酶報告基因檢測鑒定miR-17作用靶點a與R-Zfyve9-WT1+NC組比較P<0.05,b與R-Zfyve-WT1+rno-miR-17-5p組比較P<0.05,c與R-Zfyve-Mut1+rno-miR-17-5p組比較P<0.05,d與R-Zfyve-Mut2+rno-miR-17-5p組比較P<0.05
3 討論
激活素A (ActA),是轉化生長因子β(transforming growth factor-β,TGF-β)超家族的成員之一。通過ActA/Smads跨膜信號轉導,ActA在缺血性腦損傷、神經系統退行性疾病以及神經精神類疾病中均發揮神經保護作用。前期研究發現,與經典的ActA/Smads線性通路不同,ActA/Smads信號具有正反饋環路作用模式:環路活化呈細胞外ActA濃度梯度依賴,信號活化后可反過來上調ActA基因的表達[4]。這樣在外界刺激誘導ActA基因表達上調后,ActA信號可在短時間內實現跨膜信號的級聯放大和活化,繼而在SARA的輔助下通過膜受體激活Smads(R-Smads)的磷酸化活化和核內遷移,實現靶基因的表達調控,為ActA在急性腦缺血損傷的早期應答奠定生物學基礎。然而ActA/Smads環路在短時程激活后自發衰減,呈現一定的自限性[4]。盡管ActA在缺血損傷的早期及延時缺血再灌注過程中均被發現具有神經保護作用[7,8],但是受環路自限性的影響,內源性ActA信號在缺血性腦卒中早期活化后很快進入衰竭階段,將難以持續發揮生物學功能。因此,闡明ActA環路自限性及信號衰減的可能機制,并以此為切入點,尋找相應的干預靶點,延長ActA信號環路活化時程,是ActA內源性神經保護研究有待解決的關鍵問題。SARA作為TGF-β/Smads信號通路的銜接蛋白,是R-Smads向膜受體募集的重要輔助因子,具有調控R-Smads與膜受體定位,影響R-Smads平衡和分布的作用。在體外腦缺血性損傷模型中,SARA是ActA信號下游R-Smads磷酸化活化的輔助因子[9],其表達下調是ActA環路自限性的一個調控因素[4]。但引起SARA表達下調,ActA信號環路衰減的因素尚不清楚。
近期研究發現,miR-17可通過調控SARA的表達阻斷食管鱗狀細胞癌的遷移和侵襲[6]。miRNA基因轉錄的初級產物經剪切產生雙鏈RNA雙體(microRNA:microRNA*),即成熟的miRNA和其互補序列所組成的二聚體。成熟的microRNA單鏈會選擇性地整合入RISC(RNA induced silencing complex)中識別靶基因,通過mRNA剪切和抑制蛋白質翻譯的方式負性調控靶基因表達。microRNA*可能會被快速降解。大部分成熟microRNA來源于5’末端RNA鏈。本研究中的miR-17也一樣。它在包括腦組織在內的多種組織細胞廣泛表達。既往研究發現,miR-17在胚胎發育、神經元分化方面發揮作用,而且還調控了TGF-β超家族中骨形態發生蛋白(Bone morphogenetic protein,BMP)及TGF-β的信號通路轉導[10-12]。但在ActA介導的抗腦缺血損傷信號環路中,miR-17的生物學功能尚不清楚。TargetScan、miRDB檢測提示,miR-17與SARA的調控位點多物種間具有高度保守性。本研究在上述前期工作基礎上,通過實時定量PCR檢測發現miR-17在體外腦缺血性損傷后表達上調,在OGD3 h時達到高峰,隨后降低。miR-17的這種表達變化特點與既往前期研究中發現的ActA/Smads信號強度變化規律相符。進一步對miR-17功能研究發現,miR-17 mimic瞬時轉染可顯著下調SARA蛋白表達。說明miR-17可以通過調控SARA的表達水平調控ActA信號活化強度。進一步的雙熒光素酶報告基因檢測證實miR-17能夠通過這(57-64)和(403-409)位點靶向調控 SARA 表達,并且在對位點(57-64)和位點(403-409)的調控功能上呈現一定的協同作用。這樣靶向miR-17的干預可通過上調SARA表達來實現增強ActA信號的作用,并為明確ActA信號衰減機制提供數據參考。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
鴨綠江(2021年35期)2021-04-19 12:24:18
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
海峽科技與產業(2016年3期)2016-05-17 04:32:12
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
